Войти
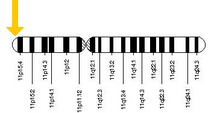 У человека ген HBB расположен на хромосоме 11 в положении p15.5.
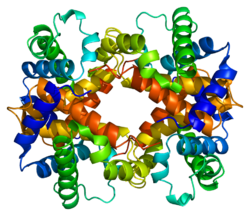
У человека ген HBB расположен на хромосоме 11 в положении p15.5. Бета-глобин (также обозначается как HBB, β-глобин, гемоглобин бета, гемоглобин бета или предпочтительно субъединица бета гемоглобина ) представляет собой белок глобин белок, который вместе с альфа-глобином (HBA ), м вызывает наиболее распространенную форму гемоглобина у взрослых людей, HbA. Он состоит из 147 аминокислот и имеет молекулярную массу 15 867 Да. Нормальный HbA взрослого человека представляет собой гетеротетрамер, состоящий из двух альфа-цепей и двух бета-цепей.
HBB кодируется геном HBB на хромосоме человека 11. Мутации в гене вызывают несколько вариантов белков, которые связаны с генетическими нарушениями, такими как серповидно-клеточная анемия и бета-талассемия, а также полезные черты, такие как генетическая устойчивость к малярии.
Белок HBB продуцируется геном HBB, который расположен в мультигенном локусе β-глобинового локуса на хромосоме 11, особенно в положении короткого плеча 15.4. Экспрессия бета-глобина и соседних глобинов в β-глобиновом локусе контролируется единственным областью контроля локуса (LCR), наиболее важным регуляторным элементом в локусе, расположенном выше глобина гены. Нормальный аллельный вариант имеет длину 1600 пар оснований (п.н.) и содержит три экзона. Порядок генов в кластере бета-глобина 5 '- эпсилон - гамма-G - гамма-A - дельта - бета - 3 '.
HBB взаимодействует с гемоглобином, альфа 1 (HBA1) с образованием гемоглобина A, основного гемоглобина в взрослые люди. Взаимодействие двоякое. Во-первых, один HBB и один HBA1 объединяются нековалентно с образованием димера. Во-вторых, два димера объединяются, образуя четырехцепочечный тетрамер, который становится функциональным гемолглобином.
Бета-талассемия является наследственной генетической мутацией. в одном (малая бета-талассемия) или обоих (большая бета-талассемия) аллелей бета-глобина на хромосоме 11. Мутантные аллели подразделяются на две группы: β0, в которой не образуется функциональный β-глобин, и β +, в которой вырабатывается небольшое количество нормального белка β-глобина. Малая бета-талассемия возникает, когда человек наследует один нормальный бета-аллель и один аномальный бета-аллель (β0 или β +). Незначительная бета-талассемия приводит к легкой микроцитарной анемии, которая часто протекает бессимптомно или может вызывать усталость или бледность кожи. Большая бета-талассемия возникает, когда человек наследует два аномальных аллеля. Это могут быть два аллеля β +, два аллеля β0 или по одному каждого из них. Большая бета-талассемия - тяжелое заболевание. Тяжелая анемия наблюдается начиная с 6-месячного возраста. Без лечения смерть часто наступает в возрасте до 12 лет. Большую бета-талассемию можно вылечить с помощью пожизненных переливаний крови или трансплантации костного мозга.
Согласно недавнему исследованию, мутация stop gain Gln40stop в гене HBB является частой причиной аутосомно-рецессивной бета-талассемии у сардинцев (почти исключительно на Сардинии). У носителей этой мутации повышенное количество эритроцитов. Любопытно, что та же мутация также была связана со снижением уровней ЛПНП в сыворотке у носителей, поэтому авторы предполагают, что это связано с необходимостью холестерина для регенерации клеточных мембран.
Было обнаружено более тысячи встречающихся в природе вариантов HBB. Наиболее распространенным является HbS, который вызывает серповидно-клеточную анемию. HbS продуцируется точечной мутацией в HBB, в которой кодон GAG заменен на GTG. Это приводит к замене гидрофильной аминокислоты глутаминовой кислоты на гидрофобную аминокислоту валин в шестом положении (β6Glu → Val). Эта замена создает гидрофобное пятно на внешней стороне белка, которое прилипает к гидрофобной области бета-цепи соседней молекулы гемоглобина. Это дополнительно вызывает скопление молекул HbS в жесткие волокна, вызывая «серповидность» всех красных кровяных телец в гомозиготном (HbS / HbS) состоянии. Гомозиготный аллель стал одним из самых смертоносных генетических факторов, тогда как люди, гетерозиготные по мутантному аллелю (HbS / HbA), устойчивы к малярии и развивают минимальные эффекты анемии.
Серповидноклеточная болезнь тесно связана с другим мутантным гемоглобином, называемым гемоглобином C (HbC), поскольку они могут передаваться вместе. Мутация HbC находится в том же положении в HbS, но глутаминовая кислота заменена на лизин (β6Glu → Lys). Мутация особенно распространена в популяциях Западной Африки. HbC обеспечивает почти полную защиту от Plasmodium falciparum у гомозиготных (CC) индивидуумов и промежуточную защиту у гетерозиготных (AC) индивидуумов. Это указывает на то, что HbC имеет более сильное влияние, чем HbS, и, по прогнозам, заменяет HbS в регионах, эндемичных по малярии.
Другая точечная мутация в HBB, в которой глутаминовая кислота заменена лизином в положении 26 (β26Glu → Lys) приводит к образованию гемоглобина E (HbE). HbE имеет очень нестабильную ассоциацию альфа- и бета-глобина. Несмотря на то, что сам по себе нестабильный белок имеет мягкий эффект, унаследованный от HbS и признаков талассемии, он превращается в опасную для жизни форму β-талассемии. Мутация возникла относительно недавно, что позволяет предположить, что она возникла в результате селективного давления против тяжелой малярии, вызванной falciparum, поскольку гетерозиготный аллель предотвращает развитие малярии.
Малярия, вызванная Plasmodium falciparum, является основным фактор отбора в эволюции человека. Он повлиял на мутации в HBB в различной степени, что привело к существованию множества вариантов HBB. Некоторые из этих мутаций не являются непосредственно летальными и вместо этого придают устойчивость к малярии, особенно в Африке, где малярия носит эпидемический характер. Люди африканского происхождения в эволюции имеют более высокий уровень мутантного HBB, потому что гетерозиготные люди имеют неправильную форму красных кровяных телец, которые предотвращают атаки малярийных паразитов. Таким образом, мутанты HBB являются источниками положительного отбора в этих регионах и важны для их длительного выживания. Такие маркеры выбора важны для отслеживания происхождения человека и диверсификации из Африки.
| Викискладе есть медиафайлы, связанные с гемоглобином, бета-цепочкой. |