
Войти
| Содержание | |
|---|---|
| Описание | Энциклопедия генов и вариантов генов |
| Типы данных. охвачены | Все особенности генов в геноме человека и мыши |
| Связаться с | |
| Исследовательским центром | Wellcome Trust Sanger Institute |
| Авторы | Харроу Дж. и др. |
| Основная ссылка | PMID 22955987 |
| Дата выпуска | сентябрь 2012 г. (сентябрь 2012 г.) |
| Доступ | |
| Веб-сайт | Генкод веб-сайта |
| Инструменты | |
| Веб | Браузер генома UCSC: http://genome.cse.ucsc.edu/encode/ |
| Разное | |
| Лицензия | Открытый доступ |
| Выпуск данных. частота | Человек - Ежеквартально. Мышь - Полугодовой |
| Версия | Человек - Выпуск 32 (сентябрь 2019 г.). Мышь - Выпуск M23 (сентябрь 2019 г.) |
GENCODE - научный проект в исследовании генома и в рамках масштабного проекта ENCODE (ENCyclopedia Of DNA Elements).
Консорциум GENCODE изначально был сформирован как часть пилотной фазы проекта ENCODE для идентификации и картирования всех генов, кодирующих белок в областях ENCODE (примерно 1% генома человека). Учитывая первоначальный успех проекта, GENCODE теперь стремится создать «Энциклопедию генов и вариантов генов», идентифицируя все особенности генов в геноме человека и мыши, используя комбинацию вычислительного анализа, ручного аннотирования и экспериментальной проверки, а также аннотируя все основанные на фактических данных особенности гена во всем геноме человека с высокой точностью.
Результатом будет набор аннотаций, включающий все кодирующие белок локусы с альтернативно транскрибируемыми вариантами, некодирующие локусы с подтверждением транскрипции и псевдогены.
GENCODE в настоящее время продвигается к своим целям в фазе 2 проекта, а именно:
Самым последним выпуском аннотаций набора генов человека является Gencode 32 с датой замораживания сентября. ber 2019. В этом выпуске используется последняя сборка эталонного генома человека GRCh38.
Последним выпуском для аннотаций генома мыши является Gencode M23, также с датой замораживания в сентябре 2019 года.
С сентября 2009 года GENCODE является набором генов человека, используемым проектом Ensembl, и каждый новый выпуск GENCODE соответствует выпуску Ensembl.
 Хронология проекта GENCODE
Хронология проекта GENCODE 2003 Сентябрь . Национальный институт исследования генома человека (NHGRI) создал общественный исследовательский консорциум под названием ENCODE, "Энциклопедия элементов ДНК" в сентябре 2003 г., чтобы осуществить проект по идентификации всех функциональных элементов в последовательности генома человека. Проект был разработан в три этапа - пилотный этап, этап разработки технологии и этап производства. Пилотная стадия проекта ENCODE была направлена на глубокое компьютерное и экспериментальное исследование 44 областей общей последовательностью 30 Мб, что составляет примерно 1% генома человека. В рамках этого этапа был сформирован консорциум GENCODE для идентификации и картирования всех генов, кодирующих белок, в регионах ENCODE. Предполагалось, что результаты первых двух этапов будут использованы для определения наилучшего пути дальнейшего анализа оставшихся 99% генома человека на экономически эффективном и всеобъемлющем этапе производства.
2005 апрель . Первый выпуск аннотации 44 регионов ENCODE был заморожен 29 апреля 2005 г. и использовался на первом семинаре ENCODE Genome Annotation Assessment Project (E-GASP). GENCODE Release 1 содержал 416 известных локусов, 26 новых (кодирующих последовательность ДНК) локусов CDS, 82 новых локуса транскриптов, 78 предполагаемых локусов, 104 процессированных псевдогена и 66 необработанных псевдогенов.
Октябрь 2005 г. . Вторая версия (выпуск 02) была заморожена 14 октября 2005 г. и содержала обновления, полученные в результате экспериментальных проверок с использованием RACE и RT-PCR техники. GENCODE Release 2 содержал 411 известных локусов, 30 новых локусов CDS, 81 новый локус транскриптов, 83 предполагаемых локуса, 104 процессированных псевдогена и 66 необработанных псевдогенов.
2007 июнь . Выводы пилотного проекта были опубликованы в июне 2007 года. Результаты подчеркнули успех пилотного проекта по созданию возможной платформы и новых технологий для характеристики функциональных элементов в геноме человека, которые открывает путь к исследованию генома.
2007 октябрь . После успешной пилотной фазы на 1% генома Wellcome Trust Sanger Institute получил грант от Национального института исследования генома человека США (NHGRI) на осуществить расширение проекта GENCODE для интегрированной аннотации генных характеристик. Это новое финансирование было частью усилий NHGRI по расширению проекта ENCODE до стадии производства для всего генома вместе с дополнительными экспериментальными исследованиями.
2012 Сентябрь . В сентябре 2012 года консорциум GENCODE опубликовал крупный документ, в котором обсуждались результаты основного выпуска - GENCODE Release 7, который был заморожен в декабре 2011 года. В выпуске GENCODE 7 использовалось сочетание руководств. аннотации генов от группы анализа и аннотаций человека и позвоночных (HAVANA) и полностью новый выпуск (Ensembl выпуск 62) автоматической аннотации генов от Ensembl. На момент выпуска GENCODE Release 7 содержал наиболее полную общедоступную аннотацию локусов длинной некодирующей РНК (днРНК) с преобладающей формой транскрипта, состоящей из двух экзонов.
2013-2017 . Участие в Группа GENCODE, успешно предоставившая полную аннотацию функциональных элементов в геноме человека, получила в 2013 году второй грант для продолжения работы по аннотации генома человека и расширения GENCODE для включения аннотации генома мыши. Предполагается, что данные аннотации мышей позволят проводить сравнительные исследования между геномами человека и мыши, чтобы улучшить качество аннотаций в обоих геномах.
Ключевые участники проекта GENCODE оставались относительно последовательными на всех его этапах, и теперь Wellcome Trust Sanger Institute возглавляет общие усилия по проекту.
Сводка основных участвующих организаций на каждой фазе приведена ниже:
| GENCODE, фаза 2 (текущая) | GENCODE, фаза расширения | GENCODE, пилотная фаза | |
|---|---|---|---|
| Wellcome Trust Sanger Institute, Кембридж, Великобритания | Wellcome Trust Sanger Institute, Кембридж, Великобритания | Wellcome Trust Sanger Institute, Кембридж, Великобритания
| |
| Центр регулирования Геномики (CRG), Барселона, Каталония, Испания | Центр регулирования Геномики (CRG), Барселона, Каталония, Испания | Institut Municipal d'Investigació Mèdica (IMIM), Барселона, Каталония, Испания | |
| Университет Лозанны, Швейцария | Университет Лозанны, Швейцария | Университет Женевы, Швейцария | |
| Калифорнийский университет, Санта-Крус (UCSC), Калифорния, США | Калифорнийский университет (UCSC), Санта-Крус, США | Вашингтонский университет (Вашингтонский университет), Сент-Луис, США | |
| Массачус etts Institute of Technology (MIT), Бостон, США | Массачусетский технологический институт (MIT), Бостон, США | Калифорнийский университет, Беркли, США | |
| Йельский университет (Йель), Нью-Хейвен, США | Йельский университет (Йель), Нью-Хейвен, США | Европейский институт биоинформатики, Хинкстон, Великобритания | |
| Испанский национальный центр исследования рака (CNIO), Мадрид, Испания | Испанский национальный центр исследований рака (CNIO), Мадрид, Испания | ||
| Вашингтонский университет (Вашингтонский университет), Сент-Луис, США |
С момента своего создания GENCODE выпустила 20 версий аннотации набора генов человека (за исключением незначительных обновлений).
Ключевая сводная статистика самой последней аннотации набора генов GENCODE Human (Release 20, апрель 2014 замораживание, Ensembl 76 ), которая является первой версией, в которой используется последняя версия Human. Сборка генома (GRCh38) показана ниже:
| Категории | Всего | Категории | Всего |
|---|---|---|---|
| Общее количество генов | 58,688 | Общее количество транскриптов | 194,334 |
| Гены, кодирующие белок | 19,942 | Транскрипты, кодирующие белок | 79,460 |
| Длинные некодирующие гены РНК | 14,470 | - полноразмерное кодирование белка: | 54,447 |
| Малые гены некодирующих РНК | 9,519 | - кодирование белка с частичной длиной: | 25,013 |
| Псевдогены | 14,363 | Транскрипты опосредованного бессмысленным распадом | 13,229 |
| - обработанные псевдогены: | 10,736 | Транскрипты длинных некодирующих РНК-локусов | 24,489 |
| - необработанные псевдогены: | 3,202 | ||
| - унитарные пс eudogenes: | 171 | ||
| - полиморфные псевдогены: | 26 | ||
| - псевдогены: | 2 | ||
| сегменты гена иммуноглобулина / рецептора Т-клеток | 618 | Общее количество различных трансляций | 59,575 |
| - сегменты, кодирующие белок: | 392 | Гены, которые имеют более одной отдельной трансляции | 13,579 |
| - псевдогены: | 226 |
См. GENCODE Statistics README и страница биотипов GENCODE для получения более подробной информации о классификации указанного выше набора генов..
Благодаря достижениям в технологиях секвенирования (например, RT-PCR-seq), расширенному охвату ручных аннотаций (группа HAVANA) и усовершенствованиям алгоритмов автоматического аннотирования с помощью Ensembl точность и полнота аннотаций GENCODE постоянно повышались. дорабатывается через итерацию выпусков.
Ниже показано сравнение ключевых статистических данных трех основных выпусков GENCODE. Очевидно, что, хотя охват, с точки зрения общего числа обнаруженных генов, неуклонно увеличивается, количество генов, кодирующих белок, фактически уменьшилось. В основном это связано с новыми экспериментальными данными, полученными с использованием кластеров Cap Analysis Gene Expression (CAGE), аннотированных сайтов PolyA и попаданий пептида.
Сравнение версий GENCODE (транскрипты)

Сравнение версий GENCODE (гены)
Сравнение версий GENCODE для людей (переводы)
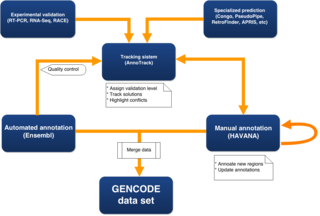 Схема конвейера GENCODE. Схема показывает поток данных между ручными и автоматизированными аннотациями через специализированные конвейеры прогнозирования, чтобы предоставить подсказки для первого прохода аннотации и контроля качества (QC). Аннотированные генные модели подлежат экспериментальной проверке, а система отслеживания AnnoTrack содержит данные из всех этих источников и используется для выделения различий, координации контроля качества и отслеживания результатов. Ручные и автоматизированные процессы аннотации создают набор данных GENCODE, а также используются для контроля качества завершенных аннотаций.
Схема конвейера GENCODE. Схема показывает поток данных между ручными и автоматизированными аннотациями через специализированные конвейеры прогнозирования, чтобы предоставить подсказки для первого прохода аннотации и контроля качества (QC). Аннотированные генные модели подлежат экспериментальной проверке, а система отслеживания AnnoTrack содержит данные из всех этих источников и используется для выделения различий, координации контроля качества и отслеживания результатов. Ручные и автоматизированные процессы аннотации создают набор данных GENCODE, а также используются для контроля качества завершенных аннотаций. Общий процесс создания аннотации для GENCODE включает ручное управление, различные вычислительные анализы и целевые экспериментальные подходы. Предполагаемые локусы могут быть проверены с помощью влажных лабораторных экспериментов, а расчетные прогнозы анализируются вручную. В настоящее время, чтобы гарантировать, что набор аннотаций охватывает весь геном, а не только области, которые были аннотированы вручную, объединенный набор данных создается с использованием ручных аннотаций из HAVANA вместе с автоматическими аннотациями из автоматически аннотированного набора генов Ensembl. Этот процесс также добавляет уникальные полноразмерные предсказания CDS из набора кодирования белков Ensembl в аннотированные вручную гены, чтобы обеспечить максимально полную и актуальную аннотацию генома.
Транскрипты Ensembl являются продуктами системы автоматической аннотации генов Ensembl (набор конвейеров аннотаций генов), называемой сборкой гена Ensembl. Все транскрипты Ensembl основаны на экспериментальных данных, и поэтому автоматизированный конвейер полагается на мРНК и белковые последовательности, депонированные в общедоступные базы данных научным сообществом. Более того, уровни 1 и 2 белка из UniProt, нетранслируемые области (UTR), гены длинной межгенной некодирующей РНК (lincRNA) (аннотированные с использованием комбинации последовательностей кДНК и регуляторных данных из проекта Ensembl), короткие некодирующие РНК (аннотированные с использованием Ансамблевые конвейеры нкРНК).
Основной подход к ручной аннотации генов заключается в аннотировании транскриптов, выровненных по геному, и использовании геномных последовательностей в качестве эталона, а не кДНК. Готовая геномная последовательность анализируется с использованием модифицированного конвейера Ensembl, а результаты BLAST для кДНК / EST и белков, наряду с различными ab initio предсказаниями, могут быть проанализированы вручную в инструменте браузера аннотаций Otterlace. Таким образом, можно предсказать больше альтернативных вариантов сплайсинга по сравнению с аннотацией кДНК. Более того, геномная аннотация дает более полный анализ псевдогенов. В консорциуме GENCODE есть несколько аналитических групп, которые запускают конвейеры, которые помогают ручным аннотаторам создавать модели в неаннотированных регионах и выявлять потенциально пропущенные или неправильные ручные аннотации, включая полностью отсутствующие локусы, отсутствующие альтернативные изоформы, неправильные места сплайсинга и неправильные биотипы. Они отправляются обратно в ручные аннотаторы с помощью системы отслеживания AnnoTrack. Некоторые из этих конвейеров используют данные из других подгрупп ENCODE, включая данные RNASeq, модификацию гистонов и данные CAGE и Ditag. Данные RNAseq - важный новый источник доказательств, но создание на их основе полных генных моделей - сложная проблема. В рамках GENCODE был проведен конкурс для оценки качества прогнозов, производимых различными конвейерами прогнозирования RNAseq (см. RGASP ниже). Для подтверждения неопределенных моделей в GENCODE также есть экспериментальный конвейер валидации с использованием секвенирования РНК и процесса слияния генов RACE
В процессе слияния сначала сравниваются все модели транскриптов HAVANA и Ensembl. путем кластеризации перекрывающихся кодирующих экзонов на одной и той же цепи, а затем путем попарного сравнения каждого экзона в кластере транскриптов. Модуль, используемый для объединения набора генов, - HavanaAdder. Перед запуском кода HavanaAdder требуются дополнительные шаги (например, система проверки работоспособности Ensembl и запросы к набору генов CCDS и выравнивания кДНК Ensembl). Если аннотация, описанная во внешних наборах данных, отсутствует в ручном наборе, то она сохраняется в системе AnnoTrack для проверки.
Для GENCODE 7 моделям транскрипции присваивается высокий или низкий уровень поддержки, основанный на новом методе, разработанном для оценки качества стенограмм. Этот метод основан на выравнивании мРНК и EST, предоставленных UCSC и Ensembl. Выравнивания мРНК и EST сравнивают с транскриптами GENCODE, и транскрипты оценивают в баллах в соответствии с выравниванием по всей длине. Сводка уровней поддержки для каждой хромосомы в GENCODE Release 7 показана на рисунке справа. Аннотации делятся на аннотации, созданные с помощью автоматизированного процесса, ручного метода и объединенные аннотации, где оба процесса приводят к одной и той же аннотации.
Усиление, упорядочение, сопоставление и проверка соединение экзон-экзон
Двухцепочечная кДНК восьми тканей человека (головной мозг, сердце, почки, семенники, печень, селезенка, легкие и скелетные мышцы) была создана с помощью амплификации кДНК, а очищенная ДНК была непосредственно использована для получения библиотеку секвенирования с помощью «Набор для подготовки образцов геномной ДНК» (Illumina). Эта библиотека была впоследствии секвенирована на платформе Illumina Genome Analyzer 2. Затем считывания (35 или 75 нт) были сопоставлены с эталонным геномом человека (hg19) и предсказанными сплайсированными ампликонами с помощью программного обеспечения Bowtie. Только считывания с уникальным отображением без несоответствия считались подтверждением сайта сплайсинга (транскрипта). Сварные стыки были подтверждены, если минимум 10 считываний со следующими характеристиками охватили прогнозируемые стыковые стыки. Для считываний длиной 35 и 75 нуклеотидов требовалось по крайней мере 4 и 8 нуклеотидов на каждой стороне контрольных точек (т.е. на каждом целевом экзоне) соответственно.
Сравнение расшифровок RefSeq, UCSC, AceView и GENCODE
Транскрипты, принадлежащие четырем различным наборам данных (GENCODE, RefSeq, UCSC и AceView), сравнивались, чтобы оценить, в какой степени эти наборы данных перекрываются. Сравнивались выпуски GENCODE 7, RefSeq и UCSC Genes freeze, июль 2011 г., и выпуск AceView 2010. Перекрытия между различными комбинациями наборов данных были графически представлены в виде трехсторонних диаграмм Венна с использованием пакета Vennerable R и отредактированы вручную.
Анализ PhyloCSF
PhyloCSF использовался для идентификации потенциальных новых кодирующих генов в РНК. -seq модели транскриптов, основанные на эволюционных сигнатурах. Для каждой модели транскрипта, созданной на основе данных Illumina HBM с использованием Exonerate или Scripture, выравнивание млекопитающих было произведено путем извлечения выравнивания каждого экзона из выравнивания UCSC позвоночных (которое включает 33 плацентарных млекопитающих).
APPRIS (CNIO)
APPRIS - это система, которая развертывает ряд вычислительных методов для придания ценности аннотациям человеческого генома. APPRIS также выбирает одну из CDS для каждого гена в качестве основной изоформы. Более того, он определяет основной вариант, объединяя структурную и функциональную информацию о белках и информацию о сохранении родственных видов. Сервер APPRIS использовался в контексте расширения проекта ENCODE для аннотирования генома человека, но APPRIS используется для других видов (например, мыши, крысы и рыбок данио). Конвейер состоит из отдельных модулей, которые объединяют информацию о структуре и функциях белка, а также данные об эволюции. Каждый модуль реализован как отдельный веб-сервис.
Текущая версия набора генов GENCODE Human (GENCODE Release 20) включает файлы аннотаций (в форматах GTF и GFF3), файлы FASTA и файлы METADATA, связанные с аннотацией GENCODE на всех геномные регионы (референсные хромосомы / участки / скаффолды / гаплотипы). Данные аннотации относятся к эталонным хромосомам и хранятся в отдельных файлах, которые включают: аннотацию генов, особенности PolyA, аннотированные HAVANA, псевдогены (Retrotransposed), предсказанные конвейерами Yale и UCSC, но не HAVANA, длинные некодирующие РНК и тРНК. структуры, предсказанные тРНК-сканированием. Некоторые примеры строк в формате GTF показаны ниже:
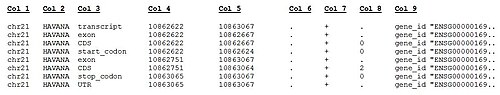 Пример файла GTF, где показаны стандартные столбцы GTF, разделенные табуляцией (1–9)
Пример файла GTF, где показаны стандартные столбцы GTF, разделенные табуляцией (1–9) Столбцы в форматах файлов GENCODE GTF описаны ниже.
Описание формата файла GENCODE GTF. Стандартные столбцы GTF, разделенные табуляцией
| Номер столбца | Содержимое | Значения / формат |
|---|---|---|
| 1 | имя хромосомы | chr {1,2,3,4,5, 6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22, X, Y, M} |
| 2 | источник аннотации | {ENSEMBL, HAVANA} |
| 3 | feature-type | {ген, транскрипт, экзон, CDS, UTR, start_codon, stop_codon, Selenocysteine} |
| 4 | место начала генома | целочисленное значение (На основе 1) |
| 5 | конечное местоположение генома | целочисленное значение |
| 6 | оценка (не используется) | . |
| 7 | геномная цепь | {+, -} |
| 8 | фаза генома ( для функций CDS) | {0,1,2,.} |
| 9 | дополнительная информация в виде пар ключ-значение | См. пояснения в таблице ниже. |
Описание пар ключ-значение в 9-м столбце файла GENCODE GTF (формат: ключ "значение")
| Имя ключа | Формат значения |
|---|---|
| gene_id | ENSGXXXXXXXXXXX |
| идентификатор_транскрипта | ENSTXXXXXXXXXXX |
| тип_гена | список биотипов |
| состояние_гена | {ИЗВЕСТНО, НОВОЕ, ПУТАТИВНОЕ} |
| имя_гена | строка |
| transcript_type | список биотипов |
| transcript_status | {KNOWN, NOVEL, PUTATIVE} |
| transcript_name | string |
| exon_number | указывает биологическое положение экзона в транскрипте |
| exon_id | ENSEXXXXXXXXXXX |
| уровень |
|
Каждый ген в наборе данных GENCODE классифицируется на три уровня в соответствии с их типом аннотации:
Уровень 1 (проверенные локусы): Включает транскрипты которые были вручную аннотированы и экспериментально подтверждены с помощью RT-PCR-seq, и псевдогенами, которые были проверены с помощью трех различных методологий.
Уровень 2 (локусы с ручными аннотациями): Выделяет стенограммы, которые были вручную аннотированы только HAVANA, а также включает стенограммы, которые были объединены с моделями, созданными автоматическим конвейером Ensembl.
Уровень 3 (локусы с автоматической аннотацией): Обозначает транскрипты и предсказания псевдогенов, полученные в результате автоматизированного конвейера аннотаций Ensembl.
Генам и транскриптам присваивается статус "известные", "новые" или "предполагаемые" в зависимости от их присутствия в других основных базах данных и доказательств, используемых для построения их составных стенограмм.
Известно: Представлен в базе данных Комитета по номенклатуре генов HUGO (HGNC) и RefSeq.
Роман: В настоящее время не представлен в базах данных HGNC или RefSeq, но хорошо подтверждается либо свидетельством транскрипта, специфичным для локуса, либо свидетельством из паралогичного или ортологичного локуса.
Предполагаемый: В настоящее время не представлен в базах данных HGNC или RefSeq, но подтвержден более короткими и более разреженными свидетельствами транскрипции.
Кроме того, на веб-сайте GENCODE есть браузер генома для человека и мыши, с помощью которого вы можете добраться до любой области генома, указав номер хромосомы и начальную конечную позицию (например, 22: 30,700,000..30,900,000), а также по идентификатору транскрипта ENS (с / без версии), идентификатору гена ENS (с / без версии) и имени гена. Браузер работает на Biodalliance.
Определение «гена» никогда не было тривиальной проблемой, с многочисленными определениями и понятиями, предложенными на протяжении многих лет с момента открытия человеческий геном. Сначала гены были задуманы в 1900-х годах как дискретные единицы наследственности, затем это считалось планом для синтеза белка, а в более позднее время его определяли как генетический код, который транскрибируется в РНК. Хотя определение гена сильно изменилось за последнее столетие, оно остается сложной и противоречивой темой для многих исследователей. С появлением проекта ENCODE / GENCODE были обнаружены еще более проблемные аспекты определения, включая альтернативный сплайсинг (при котором серии экзонов разделены интронами), межгенные транскрипции и сложные паттерны рассредоточенной регуляции, а также не -генная консервация и обилие некодирующих генов РНК. Поскольку GENCODE стремится создать энциклопедию генов и вариантов генов, эти проблемы представляли растущую проблему для проекта GENCODE по разработке обновленного понятия гена.
Псевдогены имеют ДНК последовательности, которые подобны функциональным генам, кодирующим белок, однако их транскрипты обычно идентифицируются со сдвигом рамки считывания или делецией и обычно аннотируются как побочный продукт аннотации генов, кодирующих белок в большинстве генетических баз данных. Однако недавний анализ ретротранспонированных псевдогенов обнаружил, что некоторые ретранспонированные псевдогены экспрессируются и функционируют и оказывают значительное биологическое / регуляторное воздействие на биологию человека. Чтобы справиться с неизвестными и сложностями псевдогенов, GENCODE создала онтологию псевдогенов, используя комбинацию автоматизированных, ручных и экспериментальных методов, чтобы связать различные биологические свойства, такие как особенности последовательности, эволюция и потенциальные биологические функции с псевдогенами.
Энциклопедия элементов ДНК (ENCODE) - это общественный исследовательский консорциум, созданный Национальным исследовательским институтом генома человека (NHGRI) в сентябре 2003 г. (пилотный этап). Целью ENCODE является создание исчерпывающего списка частей функциональных элементов в геноме человека, включая элементы, которые действуют на уровне белка и РНК, а также регуляторные элементы, которые контролируют клетки и условия, в которых ген активен. Анализ данных во время пилотной фазы (2003–2007 гг.) Координировался группой Ensembl, совместным проектом EBI и Wellcome Trust Sanger Institute. На начальных этапах пилотного проекта и разработки технологий 44 региона - примерно 1% генома человека - были нацелены на анализ с использованием различных экспериментальных и вычислительных методов. Все данные, полученные исследователями ENCODE, и результаты аналитических проектов ENCODE с 2003 по 2012 год хранятся в браузере и базе данных UCSC Genome. Результаты ENCODE за 2013 год и позже доступны для бесплатного скачивания и анализа на портале проекта ENCODE. Чтобы аннотировать все основанные на фактах особенности генов (гены, транскрипты, кодирующие последовательности и т. Д.) Во всем геноме человека с высокой точностью, консорциум ENCODE создает подпроект GENCODE.
Проект генома человека представлял собой международную исследовательскую работу по определению последовательности генома человека и идентификации содержащихся в нем генов. Проект координировали Национальные институты здравоохранения и Министерство энергетики США. Среди дополнительных участников были университеты в Соединенных Штатах и международные партнеры в Великобритании, Франции, Германии, Японии и Китае. Проект «Геном человека» официально начался в 1990 году и был завершен в 2003 году, на 2 года раньше запланированного срока. После обнародования полной последовательности генома человека в апреле 2003 года научное сообщество активизировало свои усилия по поиску данных, чтобы понять, как организм работает в состоянии здоровья и болезни. Основным требованием для такого понимания биологии человека является способность идентифицировать и характеризовать функциональные элементы, основанные на последовательностях, посредством экспериментов и компьютерного анализа. В сентябре 2003 года NHGRI представила проект ENCODE для облегчения идентификации и анализа полного набора функциональных элементов в последовательности человеческого генома.
Ensembl является частью проекта GENCODE, и он сыграл важную роль в обеспечении автоматической аннотации к сборке эталонного генома человека и объединении этой аннотации с ручной аннотацией от команды HAVANA. Набор генов, предоставленный Ensembl для человека, представляет собой набор генов GENCODE
Ключевой областью исследований проекта GENCODE было изучение биологической значимости длинных некодирующих РНК ( днРНК). Чтобы лучше понять экспрессию lncRNA у людей, GENCODE создал подпроект для разработки пользовательских платформ микрочипов, способных количественно определять транскрипты в аннотации lncRNA GENCODE. Ряд дизайнов был создан с использованием системы Agilent Technologies eArray, и эти дизайны доступны в стандартном пользовательском формате Agilent.
Геном RNA-seq Проект Annotation Assessment Project (RGASP) предназначен для оценки эффективности различных вычислительных методов для высококачественного анализа данных о последовательности РНК. Основные цели RGASP - обеспечить беспристрастную оценку программного обеспечения для выравнивания РНК-seq, характеристики транскрипта (обнаружение, реконструкция и количественная оценка), а также определить возможность автоматизированного аннотации генома на основе секвенирования транскриптома.
RGASP - это были организованы в рамках консорциума, смоделированного по образцу семинара по прогнозированию генов EGASP (ENCODE Genome Annotation Assessment Project), и были проведены два раунда семинаров для рассмотрения различных аспектов анализа РНК-секвенирования, а также изменения технологий и форматов секвенирования. Одним из основных открытий первого и второго раундов проекта было то, что согласование считываемых данных влияет на качество прогнозов генов. Таким образом, в настоящее время проводится третий раунд семинара RGASP (в 2014 г.), в котором основное внимание уделяется картированию считывания в геном.