
Войти
 Воспроизвести медиа Визуализация убиквитилирования
Воспроизвести медиа Визуализация убиквитилирования Ферментный катализ - это увеличение скорость процесса с помощью биологической молекулы, «фермента ». Большинство ферментов - это белки, и большинство таких процессов - это химические реакции. Внутри фермента катализ обычно происходит в локализованном месте, называемом активным центром.
. Большинство ферментов состоят преимущественно из белков, либо из одной белковой цепи, либо из множества таких цепей в многосубъединичный комплекс. Ферменты часто также включают небелковые компоненты, такие как ионы металлов или специализированные органические молекулы, известные как кофактор (например, аденозинтрифосфат ). Многие кофакторы являются витаминами, и их роль как витаминов напрямую связана с их использованием в катализе биологических процессов метаболизма. Катализ биохимических реакций в клетке имеет жизненно важное значение, поскольку многие, но не все метаболически важные реакции имеют очень низкую скорость, когда они некатализируются. Одним из драйверов эволюции белка является оптимизация такой каталитической активности, хотя только самые важные ферменты работают вблизи пределов каталитической эффективности, а многие ферменты далеки от оптимальных. Важными факторами в ферментативном катализе являются общий кислотный и основной катализ, орбитальное управление, энтропийное ограничение, эффекты ориентации (например, каталитический и ключевой катализ), а также двигательные эффекты, включающие динамику белка
Механизмы ферментативного катализа различаются, но они все они в принципе аналогичны другим типам химического катализа в том, что решающим фактором является уменьшение энергетического барьера (ов), отделяющего реагенты от продуктов. Снижение энергии активации (Ea) увеличивает долю молекул реагента, которые могут преодолеть этот барьер и образовать продукт. Важный принцип заключается в том, что, поскольку они только уменьшают энергетические барьеры между продуктами и реагентами, ферменты всегда катализируют реакции в обоих направлениях и не могут продвигать реакцию вперед или влиять на положение равновесия - только на скорость, с которой она достигается. Как и в случае с другими катализаторами, фермент не расходуется и не изменяется в результате реакции (как субстрат), а рециркулируется, так что один фермент выполняет множество циклов катализа.
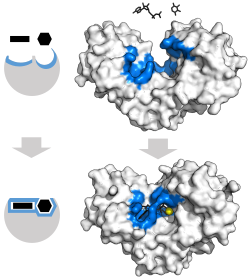 Фермент меняет форму за счет индуцированной подгонки при связывании субстрата с образуют фермент-субстратный комплекс. Гексокиназа имеет большое индуцированное движение прилегания, которое закрывает субстраты аденозинтрифосфат и ксилозу. Сайты связывания отмечены синим, субстраты - черным, а кофактор Mg - желтым. (PDB : 2E2N, 2E2Q )
Фермент меняет форму за счет индуцированной подгонки при связывании субстрата с образуют фермент-субстратный комплекс. Гексокиназа имеет большое индуцированное движение прилегания, которое закрывает субстраты аденозинтрифосфат и ксилозу. Сайты связывания отмечены синим, субстраты - черным, а кофактор Mg - желтым. (PDB : 2E2N, 2E2Q )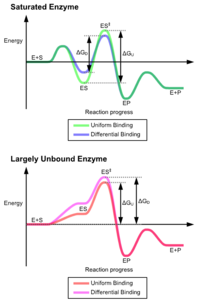 Различные механизмы связывания субстрата
Различные механизмы связывания субстрата Классической моделью взаимодействия фермент-субстрат является Модель индуцированного соответствия. Эта модель предполагает, что начальное взаимодействие между ферментом и субстратом является относительно слабым, но эти слабые взаимодействия быстро вызывают конформационные изменения в ферменте, которые усиливают связывание.
Преимущества механизма индуцированной подгонки возникают из-за стабилизирующего эффекта сильного связывания с ферментом.Существуют два различных механизма связывания субстрата: равномерное связывание, которое имеет сильное связывание субстрата, и дифференциальное связывание, которое имеет сильное связывание в переходном состоянии. связывание увеличивает аффинность связывания как субстрата, так и переходного состояния, в то время как дифференциальное связывание увеличивает только аффинность связывания в переходном состоянии. Оба они используются ферментами и эволюционно выбраны для минимизации энергии активации реакции. Ферменты th at являются насыщенными, то есть имеют высокое сродство связывания с субстратом, требуют дифференциального связывания для снижения энергии активации, тогда как небольшие несвязанные с субстратом ферменты могут использовать либо дифференциальное, либо равномерное связывание.
Эти эффекты привели к большинству белков использование механизма дифференциального связывания для снижения энергии активации, поэтому большинство субстратов имеют высокое сродство к ферменту в переходном состоянии. Дифференциальное связывание осуществляется с помощью механизма индуцированной подгонки - субстрат сначала связывается слабо, затем фермент меняет конформацию, повышая сродство к переходному состоянию и стабилизируя его, таким образом уменьшая энергию активации для его достижения.
Однако важно уточнить, что концепция индуцированной подгонки не может использоваться для рационализации катализа. То есть химический катализ определяется как уменьшение E a (когда система уже находится в ES) относительно E a в некаталитической реакции в воде (без фермента). Индуцированная подгонка только предполагает, что барьер ниже в закрытой форме фермента, но не говорит нам, какова причина снижения барьера.
Индуцированная подгонка может быть полезна для точности молекулярного распознавания в присутствии конкуренции и шума через механизм проверки конформации.
Эти конформационные изменения также приближают каталитические остатки в активном центре к химическим связям в субстрате, которые будут изменены в реакции. После того, как происходит связывание, один или несколько механизмов катализа понижают энергию переходного состояния реакции, обеспечивая альтернативный химический путь для реакции. Существует шесть возможных механизмов катализа «через барьер», а также механизм «сквозь барьер»:
Взаимодействия фермент-субстрат выравнивают реактивные химические группы и удерживают их близко друг к другу в оптимальной геометрии, что увеличивает скорость реакции. Это снижает энтропию реагентов и, таким образом, делает реакции присоединения или переноса менее неблагоприятными, так как снижение общей энтропии, когда два реагента становятся единым продуктом. Однако это общий эффект, который наблюдается в реакциях без добавления или переноса, когда он возникает из-за увеличения «эффективной концентрации» реагентов. Это понимается при рассмотрении того, как увеличение концентрации приводит к увеличению скорости реакции: по сути, когда реагенты более концентрированы, они чаще сталкиваются и поэтому реагируют чаще. При ферментативном катализе связывание реагентов с ферментом ограничивает конформационное пространство реагентов, удерживая их в `` правильной ориентации '' и близко друг к другу, так что сталкиваются чаще и с правильной геометрией, чтобы облегчить желаемая реакция. «Эффективная концентрация» - это концентрация, которой реагент должен быть в свободном состоянии в растворе, чтобы испытать ту же частоту столкновений. Часто такие теоретические эффективные концентрации нефизичны и невозможно реализовать в реальности, что является свидетельством огромной каталитической силы многих ферментов с огромным увеличением скорости по сравнению с некаталитическим состоянием.
| Например: |
| Подобные реакции будут происходить намного быстрее, если реакция является внутримолекулярной. |
 |
| Эффективная концентрация ацетата во внутримолекулярной реакции может быть оценена как k 2/k1= 2 x 10 моляр. |
Однако ситуация может быть более сложной, поскольку современные компьютерные исследования установили, что традиционные примеры эффектов близости не могут быть напрямую связаны с энтропийными эффектами ферментов. Кроме того, было обнаружено, что первоначальное предложение энтропии в значительной степени переоценивает вклад ориентационной энтропии в катализ.
Доноры и акцепторы протонов, т.е. кислоты и основание может отдавать и принимать протоны, чтобы стабилизировать развивающиеся заряды в переходном состоянии. Это связано с общим принципом катализа, принципом уменьшения энергетических барьеров, поскольку в целом переходные состояния являются состояниями с высокой энергией, и за счет их стабилизации эта высокая энергия снижается, понижая барьер. Ключевой особенностью ферментного катализа по сравнению с многими другими видами катализа небиологического происхождения является то, что и кислотный, и основной катализ могут быть объединены в одной реакции. Во многих абиотических системах кислоты (большие [H +]) или основания (стоки H + с большой концентрацией или разновидности с электронными парами) могут увеличивать скорость реакции; но, конечно, среда может иметь только один общий pH (показатель кислотности или щелочности (щелочности)). Однако, поскольку ферменты представляют собой большие молекулы, они могут позиционировать как кислотные группы, так и основные группы в своем активном центре для взаимодействия со своими субстратами, и использовать оба режима независимо от общего pH.
Часто используется общий кислотный или основной катализ для активации нуклеофильных и / или электрофильных групп или для стабилизации уходящих групп. Многие аминокислоты с кислотными или основными группами используются в активном центре, такие как глутаминовая и аспарагиновая кислоты, гистидин, цистин, тирозин, лизин и аргинин, а также серин и треонин. Кроме того, часто используется пептидный каркас с карбонильными и амидными N-группами. Цистин и гистидин используются очень часто, поскольку оба они имеют pKa, близкое к нейтральному pH, и поэтому могут как принимать, так и отдавать протоны..
Многие механизмы реакции, включающие кислотно-щелочной катализ, предполагают существенно измененное pKa. Это изменение pKa возможно через локальное окружение остатка.
| Условия | Кислоты | Основания |
|---|---|---|
| Гидрофобная среда | Увеличить pKa | Уменьшить pKa |
| Соседние остатки с одинаковым зарядом | Увеличить pKa | Уменьшить pKa |
| Образование солевого мостика (и водородной. связи) | Уменьшить pKa | Увеличить pKa |
pKa также можно находится под значительным влиянием окружающей среды до такой степени, что остатки, которые являются основными в растворе, могут действовать как доноры протонов, и наоборот.
| Например: |
| Каталитическая триада сериновой протеазы |
 |
| Начальная стадия каталитического механизма сериновой протеазы включает гистидин активного центра, принимающий протон от серинового остатка. Это подготавливает серин как нуклеофил для атаки амидной связи субстрата. Этот механизм включает передачу протона от серина (основание, pKa 14) гистидину (кислота, pKa 6), что стало возможным благодаря локальному окружению оснований. |
Важно уточнить, что модификация pKa является чистой частью электростатического механизма. Кроме того, каталитический эффект приведенного выше примера в основном связан с уменьшением pKa оксианиона и увеличением pKa гистидина, в то время как перенос протона от серина к гистидину не катализируется в значительной степени, поскольку он не является барьер, определяющий скорость.
Стабилизация заряженных переходных состояний также может происходить за счет остатков в активном центре, образующих ионные связи (или взаимодействия частичного ионного заряда) с промежуточный. Эти связи могут происходить из кислотных или основных боковых цепей, обнаруженных в аминокислотах, таких как лизин, аргинин, аспарагиновая кислота или глутаминовая кислота, или происходят из металлических кофакторов, таких как цинк. Ионы металлов особенно эффективны и могут снизить pKa воды в достаточной степени, чтобы сделать ее эффективным нуклеофилом.
Систематические исследования с компьютерным моделированием показали, что электростатические эффекты, безусловно, вносят наибольший вклад в катализ. Он может увеличить скорость реакции до 10 раз. В частности, было обнаружено, что фермент обеспечивает более полярную среду, чем вода, и что ионные переходные состояния стабилизируются фиксированными диполями. Это сильно отличается от стабилизации переходного состояния в воде, где молекулы воды должны платить «энергией реорганизации». Для стабилизации ионного и заряженного состояний. Таким образом, катализ связан с тем фактом, что полярные группы фермента предварительно организованы
Было показано, что величина электростатического поля, создаваемого активным центром фермента, сильно коррелирует с повышением каталитической скорости фермента
Связывание субстрата обычно исключает попадание воды в активный центр, тем самым снижая локальную диэлектрическую постоянную до диэлектрической проницаемости органического растворителя. Это усиливает электростатические взаимодействия между заряженными / полярными подложками и активными центрами. Кроме того, исследования показали, что распределения заряда вокруг активных центров устроены таким образом, чтобы стабилизировать переходные состояния катализированных реакций. В некоторых ферментах эти распределения заряда, по-видимому, служат для направления полярных субстратов к их сайтам связывания, так что скорости этих ферментативных реакций превышают их очевидные пределы, контролируемые диффузией.
| Например: |
| Карбоксипептидаза каталитический механизм |
 |
| Тетраэдрический промежуточный продукт стабилизируется частичной ионной связью между ионом Zn и отрицательным зарядом кислорода. |
Ковалентный катализ включает в себя образование субстратом временной ковалентной связи с остатками в активном центре фермента или с кофактором. Это добавляет к реакции дополнительный ковалентный промежуточный продукт и помогает снизить энергию более поздних переходных состояний реакции. Ковалентная связь должна быть разорвана на более поздней стадии реакции для регенерации фермента. Этот механизм используется каталитической триадой ферментов, таких как протеазы, такие как химотрипсин и трипсин, где образуется промежуточный ацил-фермент.. Альтернативным механизмом является образование основания Шиффа с использованием свободного амина из остатка лизина, как показано в ферменте альдолазе во время гликолиз.
Некоторые ферменты используют не аминокислотные кофакторы, такие как пиридоксальфосфат (PLP) или тиаминпирофосфат (TPP), для образования ковалентных промежуточных продуктов с молекулами реагентов. Такие ковалентные промежуточные соединения действуют для снижения энергии более поздних переходных состояний, подобно тому, как ковалентные промежуточные соединения, образованные с помощью аминокислотных остатков активного центра, обеспечивают стабилизацию, но возможности кофакторов позволяют ферментам проводить реакции, которые не могут выполнять только боковые аминокислотные остатки. Ферменты, использующие такие кофакторы, включают PLP-зависимый фермент аспартаттрансаминазу и TPP-зависимый фермент пируватдегидрогеназа.
Вместо того, чтобы снижать энергию активации для пути реакции, ковалентный катализ предоставляет альтернативу путь реакции (через ковалентный промежуточный продукт) и поэтому отличается от истинного катализа. Например, энергетику ковалентной связи с молекулой серина в химотрипсине следует сравнивать с хорошо изученной ковалентной связью с нуклеофилом в реакции некаталитического раствора. Истинное предложение ковалентного катализа (где барьер ниже, чем соответствующий барьер в растворе) потребует, например, частичной ковалентной связи с переходным состоянием группой фермента (например, очень прочной водородной связи) и т. эффекты не вносят значительного вклада в катализ.
Ион металла в активном центре участвует в катализе, координируя стабилизацию заряда и экранирование. Из-за положительного заряда металла только отрицательные заряды могут быть стабилизированы ионами металлов. Однако ионы металлов полезны в биологическом катализе, потому что на них не влияют изменения pH. Ионы металлов также могут ионизировать воду, действуя как кислота Льюиса. Ионы металлов также могут быть агентами окисления и восстановления.
Это основной эффект индуцированного связывания, когда сродство фермента к переходному состоянию больше, чем к сам субстрат. Это вызывает структурные перестройки, которые деформируют субстрат, связывая его в положение, более близкое к конформации переходного состояния, таким образом уменьшая разницу энергий между субстратом и переходным состоянием и помогая катализировать реакцию.
Однако эффект деформации фактически является эффектом дестабилизации основного состояния, а не эффектом стабилизации переходного состояния. Кроме того, ферменты очень гибкие и не могут оказывать сильного деформационного эффекта.
Помимо деформации связи в субстрате, внутри самого фермента может также индуцироваться деформация связи для активации остатков в активном центре.
| Например: |
| Конформации субстрата, связанного субстрата и переходного состояния лизоцима. |
| Субстрат при связывании искажается из конформации полукресла гексозного кольца (из-за стерических препятствий с аминокислоты белка, заставляющие экваториальный с6 находиться в аксиальном положении) в конформацию кресла |
Эти традиционные механизмы «через барьер» в некоторых случаях были поставлены под сомнение с помощью моделей и наблюдений механизмы «сквозь барьер» (квантовое туннелирование ). Некоторые ферменты работают с кинетикой, которая быстрее, чем можно было бы предсказать с помощью классической ΔG. В моделях «сквозь барьер» протон или электрон могут туннелировать через активационные барьеры. Квантовое туннелирование протонов наблюдалось при окислении триптамина с помощью ароматической аминдегидрогеназы.
Квантовое туннелирование, по-видимому, не дает большого каталитического преимущества, поскольку вклады в туннелирование одинаковы в катализируемом и некатализируемом реакции в растворе. Однако вклад туннелирования (как правило, повышение констант скорости в ~ 1000 раз по сравнению со скоростью реакции для классического пути «через барьер»), вероятно, имеет решающее значение для жизнеспособности биологических организмов. Это подчеркивает общую важность туннельных реакций в биологии.
В 1971-1972 годах была сформулирована первая квантово-механическая модель ферментного катализа.
Энергия связи фермент-субстратного комплекса не может рассматриваться как внешняя энергия, необходимая для активации субстрата. Фермент с высоким содержанием энергии может сначала перенести некоторую конкретную энергетическую группу X 1 с каталитического сайта фермента на конечное место первого связанного реагента, а затем другую группу X 2 из второй связанный реагент (или из второй группы единственного реагента) должен быть перенесен в активный центр для завершения превращения субстрата в продукт и регенерации фермента.
Мы можем представить всю ферментативную реакцию как две реакции сочетания:
 | (1) |
 | (2) |
Из реакции (1) видно, что группа X 1 активного фермента появляется в продукте из-за возможности реакции обмена внутри фермента, чтобы избежать как электростатического торможения, так и отталкивания атомов. Таким образом, мы представляем активный фермент как мощный реагент ферментативной реакции. Реакция (2) показывает неполное превращение субстрата, поскольку его группа X 2 остается внутри фермента. Этот подход как идея ранее предлагался, полагаясь на гипотетические чрезвычайно высокие ферментативные превращения (каталитически совершенный фермент).
Ключевым моментом для проверки настоящего подхода является то, что катализатор должен быть комплексом фермента с передаточная группа реакции. Этот химический аспект подтверждается хорошо изученными механизмами нескольких ферментативных реакций. Рассмотрим реакцию гидролиза пептидной связи, катализируемую чистым протеином α-химотрипсином (фермент, действующий без кофактора), который является хорошо изученным членом семейства сериновых протеаз, см.
Мы представляем экспериментальные результаты для этой реакции в виде двух химических стадий:
 | (3) |
 | (4) |
где S 1 представляет собой полипептид, P 1 и P 2 представляют собой продукты. Первая химическая стадия (3) включает образование ковалентного промежуточного соединения ацил-фермента. Вторая стадия (4) - это стадия деацилирования. Важно отметить, что группа H +, первоначально обнаруженная на ферменте, но не в воде, появляется в продукте до стадии гидролиза, поэтому ее можно рассматривать как дополнительную группу ферментативной реакции.
Таким образом, реакция (3) показывает, что фермент действует как мощный реагент реакции. Согласно предложенной концепции, транспорт H из фермента способствует первому превращению реагента, разрыву первой начальной химической связи (между группами P 1 и P 2). Стадия гидролиза приводит к разрыву второй химической связи и регенерации фермента.
Предлагаемый химический механизм не зависит от концентрации субстратов или продуктов в среде. Однако изменение их концентрации в основном вызывает изменения свободной энергии на первой и последней стадиях реакций (1) и (2) из-за изменений содержания свободной энергии каждой молекулы, будь то S или P, в водном растворе. Этот подход соответствует следующему механизму сокращения мышц. Заключительным этапом гидролиза АТФ в скелетных мышцах является высвобождение продукта, вызванное ассоциацией миозиновых головок с актином. Закрытие актин-связывающей щели во время реакции ассоциации структурно связано с открытием нуклеотид-связывающего кармана на активном сайте миозина.
Примечательно, что заключительные стадии гидролиза АТФ включают быстрое высвобождение фосфата. и медленное высвобождение АДФ. Высвобождение фосфатного аниона из связанного аниона АДФ в водный раствор можно рассматривать как экзергоническую реакцию, поскольку фосфатный анион имеет низкую молекулярную массу.
Таким образом, мы приходим к выводу, что первичное высвобождение неорганического фосфата H 2PO4приводит к преобразованию значительной части свободной энергии гидролиза АТФ в кинетическую энергию сольватированного фосфата с образованием активного потоковое. Это предположение о локальной механохимической трансдукции согласуется с механизмом сокращения мышц Тироша, в котором сила мышц возникает из интегрированного действия активного потока, создаваемого гидролизом АТФ.
В действительности, большинство ферментных механизмов включает комбинацию нескольких различных типов катализа.
Триозофосфатизомераза (EC 5.3.1.1 ) катализирует обратимое взаимное превращение двух триозных фосфатов изомеров дигидроксиацетонфосфат и D- глицеральдегид-3-фосфат.
трипсин (EC 3.4.21.4 ) представляет собой сериновую протеазу, которая расщепляет белок субстратов после остатков лизина или аргинина с использованием каталитической триады для выполнения ковалентного катализа и оксианионной дыры для стабилизации накопления заряда в переходных состояниях .
альдолаза (EC 4.1.2.13 ) катализирует расщепление фруктозо-1,6-бисфосфата (F-1,6-BP) на глицеральдегид-3-фосфат и дигидроксиацетонфосфат (DHAP ).
Появление исследований одиночных молекул привело в 2010-х годах к наблюдению, что движение несвязанных ферментов увеличивается с увеличением концентрации субстрата и увеличением энтальпия реакции. Последующие наблюдения предполагают, что это увеличение диффузии вызвано временным смещением центра масс фермента, что приводит к «эффекту отдачи, который продвигает фермент».
Сходство между ферментативными реакциями (EC ) можно рассчитать, используя изменения связей, реакционные центры или показатели субструктуры (EC-BLAST ).