
Войти
| Реакция Дильса – Альдера | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Названа в честь | Отто Пауля Германа Дильса. Курта Алдера | ||||||||
| Реакция тип | циклоприсоединение | ||||||||
| Реакция | |||||||||
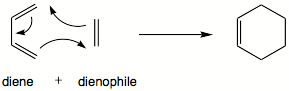 . .
| |||||||||
| Идентификаторы | |||||||||
| Портал органической химии | реакция Дильса-Альдера | ||||||||
| RSC ID онтологии | RXNO: 0000006 | ||||||||
В органической химии реакция Дильса-Альдера является химическим реакция между сопряженным диеном и замещенным алкеном, обычно называемым диенофилом (также пишется диенеофилом), с образованием замещенного производное циклогексена. Это типичный пример перициклической реакции с согласованным механизмом. Более конкретно, оно классифицируется как термически разрешенное [4 + 2] циклоприсоединение с символом Вудворда – Хоффмана [π4s+ π2s]. Впервые ее описали Отто Дильс и Курт Альдер в 1928 году. За открытие этой реакции они были удостоены Нобелевской премии по химии в 1950 году. одновременное построение двух новых углерод-углеродных связей, реакция Дильса-Альдера обеспечивает надежный способ образования шестичленных колец с хорошим контролем регио- и стереохимических результатов. Следовательно, он послужил мощным и широко применяемым инструментом для введения химической сложности в синтез природных продуктов и новых материалов. Основная концепция также была применена к π-системам, включающим гетероатомы, такие как карбонилы и имины, которые образуют соответствующие гетероциклы ; этот вариант известен как гетеро-реакция Дильса – Альдера . Реакция также была обобщена на другие размеры кольца, хотя ни одно из этих обобщений не соответствовало образованию шестичленных колец с точки зрения объема или универсальности. Из-за отрицательных значений ΔH ° и ΔS ° для типичной реакции Дильса-Альдера микроскопическая обратная реакция реакции Дильса-Альдера становится благоприятной при высоких температурах, хотя это имеет синтетическое значение только для ограниченного диапазона аддуктов Дильса-Альдера., как правило, с некоторыми особенностями конструкции; эта обратная реакция известна как ретро-реакция Дильса – Альдера.
Реакция является примером согласованной перициклической реакции. Считается, что это происходит через одно циклическое переходное состояние, без образования промежуточных продуктов в ходе реакции. Таким образом, реакция Дильса-Альдера регулируется соображениями орбитальной симметрии: она классифицируется как [π4s+ π2s] циклоприсоединение, что указывает на то, что она протекает через надфасциальное / надфазное взаимодействие 4π-электронной системы ( диеновая структура) с 2π-электронной системой (диенофильная структура), взаимодействие, которое приводит к переходному состоянию без дополнительного энергетического барьера, обусловленного орбитальной симметрией, и позволяет относительно легко протекать реакции Дильса-Альдера.
Рассмотрение пограничных молекулярных орбиталей (FMO) реагентов проясняет, почему это так. (Такой же вывод можно сделать из диаграммы орбитальной корреляции или анализа Дьюара-Циммермана.) Для более распространенной реакции Дильса-Альдера «нормального» электронного спроса более важным из двух взаимодействий HOMO / LUMO является взаимодействие между электронами и электронами. ψ 2 богатого диена в качестве наивысшей занятой молекулярной орбитали (ВЗМО) с π * электронно-дефицитного диенофила в качестве самой низкой незанятой молекулярной орбитали (НСМО). Однако энергетическая щель HOMO – LUMO достаточно близка, чтобы их роли можно было поменять местами, переключая электронные эффекты заместителей на два компонента. В обратной (обратной) реакции Дильса-Альдера с потребностью в электронах электроноакцепторные заместители на диене понижают энергию его пустых ψ 3 орбитальных и электронодонорных заместителей на диенофиле. увеличить энергию его заполненной π-орбитали настолько, чтобы взаимодействие между этими двумя орбиталями стало наиболее энергетически значимым стабилизирующим орбитальным взаимодействием. Независимо от того, какая ситуация имеет место, HOMO и LUMO компонентов находятся в фазе, и возникает скрепляющее взаимодействие, как это видно на диаграмме ниже. Поскольку реагенты находятся в основном состоянии, реакция инициируется термически и не требует активации светом.
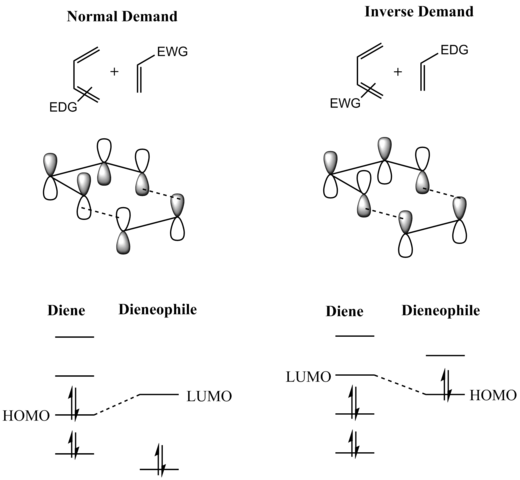
«Преобладает мнение», что большинство реакций Дильса-Альдера протекает по согласованному механизму; этот вопрос, однако, был тщательно оспорен. Несмотря на то, что подавляющее большинство реакций Дильса-Альдера демонстрируют стереоспецифическое син-добавление двух компонентов, бирадикальный промежуточный продукт был постулирован (и подтвержден расчетными данными) на том основании, что наблюдаемая стереоспецифичность не исключает двухэтапного добавление, включающее промежуточное соединение, которое разрушается до продукта быстрее, чем оно может вращаться, чтобы обеспечить инверсию стереохимии.
При проведении определенных реакций Дильса – Альдера в полярных органических растворителях, таких как диметилформамид и этиленгликоль, скорость реакции значительно увеличивается. и даже в воде. Например, реакция циклопентадиена и бутенона в воде протекает в 700 раз быстрее по сравнению с 2,2,4-триметилпентаном в качестве растворителя. Было предложено несколько объяснений этого эффекта, таких как увеличение эффективной концентрации из-за гидрофобной упаковки или стабилизации переходного состояния водородными связями.
Геометрия диеновых и диенофильных компонентов, каждая, распространяется на стереохимические детали продукт. В частности, для межмолекулярных реакций предпочтительное позиционное и стереохимическое соотношение заместителей двух компонентов по сравнению друг с другом регулируются электронными эффектами. Однако для внутримолекулярных реакций циклоприсоединения Дильса-Альдера конформационная стабильность структуры переходное состояние может иметь подавляющее влияние.
Пограничная теория молекулярных орбиталей также использовалась для объяснения закономерностей региоселективности, наблюдаемых в реакциях Дильса – Альдера замещенных систем. Расчет энергетических и орбитальных коэффициентов граничных орбиталей компонентов дает картину, которая хорошо согласуется с более прямым анализом резонансных эффектов заместителей, как показано ниже.
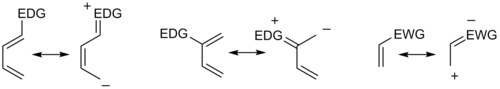
В общем, региоселективность, обнаруженная как для нормальной, так и для обратной реакции Дильса-Альдера с электронным спросом, следует так называемому орто-параправилу, потому что циклогексеновый продукт имеет заместители в положениях, аналогичных положению орто- и пара-положения дизамещенных аренов. Например, в сценарии нормального спроса диен, несущий электронодонорную группу (EDG) в C1, имеет наибольший коэффициент HOMO в C4, в то время как диенофил с электронодонорной группой (EWG) в C1 имеет наибольший коэффициент LUMO при C2. Объединение этих двух коэффициентов дает "орто" произведение, как показано в случае 1 на рисунке ниже. Диен, замещенный у С2, как в случае 2 ниже, имеет самый большой коэффициент ВЗМО у С1, давая "пара" продукт. Аналогичный анализ для соответствующих сценариев обратного спроса приводит к аналогичным продуктам, как показано в случаях 3 и 4. Изучая канонические мезомерные формы выше, легко убедиться, что эти результаты соответствуют ожиданиям, основанным на рассмотрении электронной плотности и поляризация.

В целом, что касается наиболее энергетически согласованной пары ВЗМО-НСМО, максимизация энергии взаимодействия за счет образования связей между центрами с наибольшими граничными орбитальными коэффициентами позволяет прогнозировать основной региоизомер, который будет результатом данного диен- сочетание диенофилов. При более сложной обработке три типа заместителей (Z удаление: снижение HOMO и LUMO (CF 3, NO 2, CN, C (O) CH 3), X пожертвование: повышение HOMO и LUMO (Me, OMe, NMe 2), C конъюгирование: повышение HOMO и LUMO понижение (Ph, винил)), в результате чего получается 18 возможных комбинаций. Максимизация орбитального взаимодействия правильно предсказывает продукт во всех случаях, для которых доступны экспериментальные данные. Например, в необычных комбинациях, включающих группы X как на диене, так и на диенофиле, может быть предпочтительна схема 1,3-замещения, что не объясняется аргументом упрощенной резонансной структуры. Однако случаи, когда аргумент резонанса и согласование наибольших орбитальных коэффициентов не совпадают, редки.
Реакции Дильса – Альдера, как согласованные циклоприсоединения, стереоспецифичны. Стереохимическая информация о диене и диенофиле сохраняется в продукте как син-добавка по отношению к каждому компоненту. Например, заместители в цис (транс, соответственно) отношениях на двойной связи диенофила приводят к появлению заместителей, которые являются цис (транс, соответственно) у тех же атомов углерода по отношению к циклогексеновому кольцу. Аналогичным образом, цис-, цис- и транс-дизамещенные диены дают цис-заместители у этих атомов углерода продукта, тогда как цис-, транс-дизамещенные диены дают транс-заместители:

 Эндо- и экзопереходные состояния для циклопентадиена при добавлении к акролеину ; Соотношение продуктов эндо / экзо для этого и различных других диенофилов
Эндо- и экзопереходные состояния для циклопентадиена при добавлении к акролеину ; Соотношение продуктов эндо / экзо для этого и различных других диенофилов . Реакции Дильса-Альдера, в которых соседние стереоцентры генерируются на двух концах вновь образованных одинарных связей, подразумевают два различных возможных стереохимических результата. Это стереоселективная ситуация, основанная на относительной ориентации двух отдельных компонентов, когда они реагируют друг с другом. В контексте реакции Дильса-Альдера переходное состояние, в котором наиболее значимый заместитель (электроноакцепторная и / или сопрягающая группа) диенофила ориентирован в сторону π-системы диена и скользит под ней по мере протекания реакции, называется известное как состояние эндо-перехода. В альтернативном экзопереходном состоянии он ориентирован от него. (Существует более общее использование терминов эндо и экзо в стереохимической номенклатуре.)
В случаях, когда диенофил имеет один электроноакцепторный / сопрягающий заместитель или два электроноакцепторных заместителя. / конъюгирования цис-заместителей друг с другом, результат часто можно предсказать. В этих сценариях Дильса-Альдера «нормального спроса» обычно предпочтительнее эндо-переходное состояние, несмотря на то, что оно часто более стерически перегружено. Это предпочтение известно как правило эндометрия из ольхи . Как первоначально заявил Альдер, предпочтительным переходным состоянием является состояние с «максимальным накоплением двойных связей». Эндоселективность обычно выше для жестких диенофилов, таких как малеиновый ангидрид и бензохинон ; для других, таких как акрилаты и кротонаты, селективность не очень выражена.
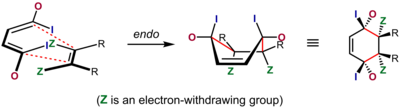
Наиболее широко распространенным объяснением происхождения этого эффекта является благоприятное взаимодействие между π-системами диенофил и диен, взаимодействие, описываемое как вторичный орбитальный эффект, хотя диполярное и ван-дер-ваальсово притяжения также могут играть определенную роль, а растворитель иногда может иметь существенное значение для избирательность. Объяснение вторичного орбитального перекрытия было впервые предложено Вудвордом и Хоффманном. В этом объяснении орбитали, связанные с группой в конъюгации с двойной связью диенофила, перекрываются с внутренними орбиталями диена, ситуация, которая возможна только для эндо-переходного состояния. Хотя в первоначальном объяснении использовалась только орбиталь на атоме α к диенофильной двойной связи, Салем и Хоук впоследствии предположили, что обе орбитали на α- и β-атомах углерода участвуют, когда позволяет молекулярная геометрия.

Часто, как и в случае сильно замещенных диенов, очень объемные диенофилы или обратимые реакции (как в случае фурана в виде диена) стерические эффекты могут преобладать над нормальной эндоселективностью в пользу экзо-изомера.
Диеновый компонент реакции Дильса-Альдера может быть либо с открытой цепью, либо циклическим, и он может содержать много различных типов заместителей; однако он должен быть способен существовать в s-цис-конформации, поскольку это единственный конформер, который может участвовать в реакции. Хотя бутадиены обычно более стабильны в s-транс-конформации, в большинстве случаев разница в энергии невелика (~ 2–5 ккал / моль).
Объемный заместитель в положении C2 или C3 может увеличить скорость реакции на дестабилизация s-транс-конформации и принуждение диена к реактивной s-цис-конформации. Например, 2-трет-бутил-бута-1,3-диен в 27 раз более реакционноспособен, чем простой бутадиен. И наоборот, диен, имеющий объемные заместители как у C2, так и у C3, менее реакционноспособен, поскольку стерические взаимодействия между заместителями дестабилизируют s-цис-конформацию.
Диены с объемными концевыми заместителями (C1 и C4) снижают скорость реакции, предположительно, препятствуя сближению диена и диенофила.
Особенно реакционноспособным диеном является 1-метокси-3-триметилсилоксибута-1,3-диен, иначе известный как диен Данишефского. Он имеет особую синтетическую полезность в качестве средства получения α, β-ненасыщенных циклогексеноновых систем путем удаления 1-метоксизаместителя после снятия защиты с енолсилилового эфира. Другие синтетически полезные производные диена Данишефского включают 1,3-алкокси-1-триметилсилокси-1,3-бутадиены (диены Брассарда) и 1-диалкиламино-3-триметилсилокси-1,3-бутадиены (диены Равала). Повышенная реакционная способность этих и подобных диенов является результатом синергетического вклада донорных групп в С1 и С3, повышая HOMO значительно выше, чем у сопоставимого монозамещенного диена.

Нестабильные (и, следовательно, очень реактивные) диены, из которых, возможно, наиболее синтетически полезными являются о- хинодиметаны, которые могут быть получены in situ. Сильная движущая сила для [4 + 2] циклоприсоединения таких видов является результатом установления (или восстановления) ароматичности. Обычные методы получения о-хинодиметанов включают пиролиз бензоциклобутенов или соответствующего сульфона, 1,4-элиминирование орто-бензилсиланов или станнанов и восстановление α, α'-ортобензильных дибромидов.
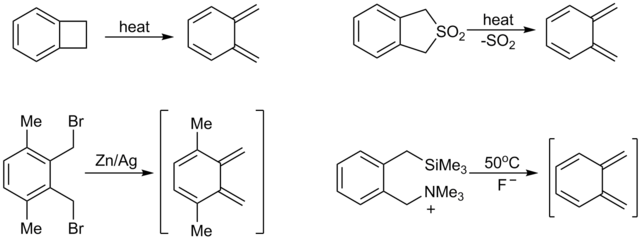
Напротив, стабильные диены представляют собой довольно инертный и вступает в реакции Дильса-Альдера только при повышенных температурах: например, нафталин может действовать как диен, приводя к аддуктам только с высокореактивными диенофилами, такими как N-фенил- малеимид. Антрацен, будучи менее ароматным (и, следовательно, более реактивным для синтезов Дильса-Альдера) в своем центральном кольце, может образовывать аддукт 9,10 с малеиновым ангидридом при 80 ° C и даже с ацетилен, слабый диенофил, при 250 ° C.
При нормальной реакции Дильса-Альдера диенофил имеет электроноакцепторную группу в сочетании с алкен; в сценарии обратной потребности диенофил конъюгирован с электронодонорной группой. Можно выбрать диенофилы, содержащие «замаскированную функциональность». Диенофил подвергается реакции Дильса-Альдера с диеном, вводя такую функциональность в молекулу продукта. Затем следует серия реакций, чтобы преобразовать функциональность в желаемую группу. Конечный продукт не может быть получен за одну стадию DA, потому что эквивалентный диенофил либо нереактивен, либо недоступен. Примером такого подхода является использование α-хлоракрилонитрила (CH 2 = CClCN). При взаимодействии с диеном этот диенофил вводит α-хлорнитрильную функциональную группу в молекулу продукта. Это «замаскированная функциональность», которая затем может быть гидролизована с образованием кетона. α-Хлоракрилонитрилдиенофил является эквивалентом кетендиенофила (CH 2 = C = O), который дает такой же продукт на одной стадии DA. Проблема в том, что сам кетен не может быть использован в реакциях Дильса-Альдера, потому что он реагирует с диенами нежелательным образом (посредством [2 + 2] циклоприсоединения), и поэтому необходимо использовать подход «замаскированной функциональности». К другим таким функциональным группам относятся фосфониевые заместители (дающие экзоциклические двойные связи после реакции Виттига), различные сульфоксидные и сульфонильные группы (обе являются эквивалентами ацетилена) и нитрогруппы (эквиваленты кетена).
Реакции Дильса-Альдера с участием по крайней мере одного гетероатома также известны и в совокупности называются гетеро-реакциями Дильса-Альдера. Карбонил группы, например, могут успешно взаимодействовать с диенами с образованием дигидропирановых колец, реакция, известная как оксо-реакция Дильса – Альдера, и иминов может использоваться либо в качестве диенофила, либо в различных участках диена для образования различных N-гетероциклических соединений посредством реакции аза-Дильса – Альдера. Нитрозосоединения (R-N = O) могут реагировать с диенами с образованием оксазинов. Хлорсульфонилизоцианат можно использовать в качестве диенофила для получения лактама Винса.
кислот Льюиса, таких как хлорид цинка, трифторид бора, тетрахлорид олова, хлорид алюминия и т. Д. y может действовать как катализатор реакций Дильса-Альдера нормальной потребности за счет координации с диенофилом. Комплексный диенофил становится более электрофильным и более реактивным по отношению к диену, увеличивая скорость реакции и часто также улучшая регио- и стереоселективность. Кислотный катализ Льюиса также позволяет реакциям Дильса-Альдера протекать при низких температурах, то есть без термической активации.
Для влияния на стереоселективность реакции Дильса-Альдера было разработано множество методов. реакции, такие как использование хиральных вспомогательных веществ, катализ хиральными кислотами Льюиса и катализаторами на малых органических молекулах. оксазолидиноны Эванса, оксазаборолидины, бис-оксазолин –медь хелаты, имидазолин катализ и многие другие методологии существуют для осуществления диастерео- и энантиоселективных реакций Дильса-Альдера.
В гексадегидро реакции Дильса – Альдера вместо алкенов используются алкины и диины и диены, образующие нестабильное промежуточное соединение бензин, которое затем можно улавливать с образованием ароматического продукта. Эта реакция позволяет образовывать сильно функционализированные ароматические кольца за одну стадию.
Ретро-реакция Дильса-Альдера используется в промышленном производстве циклопентадиена. Циклопентадиен является предшественником различных норборненов, которые являются обычными мономерами. Реакция Дильса – Альдера также используется при производстве витамина B6.
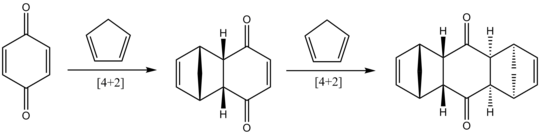 Реакция, обнаруженная Дильсом и Альдером в 1928 году.
Реакция, обнаруженная Дильсом и Альдером в 1928 году. Работа Дильса и Альдера описана в серии из 28 статей, опубликованных в Justus Liebigs Annalen der Chemie и Berichte der deutschen chemischen Gesellschaft с 1928 по 1937 год. Первые 19 статей были написаны Дильсом и Альдером, в то время как более поздние статьи были автором Дильса и различных авторов.
Первое применение реакции Дильса-Альдера в полный синтез был проиллюстрирован R. Б. Вудворд синтезирует стероиды кортизон и холестерин. Взаимодействие бутадиена с хиноном, указанным ниже, дает кольцам C и D стероидного скелета желаемую региохимию.

Э. Дж. Кори в своем первоначальном синтезе простагландинов F2α и E2 в 1969 году использовал реакцию Дильса-Альдера на ранней стадии синтеза, чтобы установить относительную стереохимию трех смежных стереоцентров на простагландин-циклопентановом ядре. Для смягчения изомеризации замещенного циклопентадиена посредством 1,5-гидридного сдвига было обнаружено, что необходимо поддерживать этот промежуточный продукт при температуре ниже 0 ° C до тех пор, пока не произойдет процесс Дильса-Альдера. Таким образом, для протекания реакции требовалась активация сильно кислым тетрафторборатом меди по Льюису. Использование 2-хлоракрилонитрила в качестве диенофила является жизнеспособным синтетическим эквивалентом кетена, структуры, которая обычно не участвует в реакции [2 + 2] циклоприсоединения с образованием димера циклобутанона, а не участвует в реакциях Дильса-Альдера с 1, 3-диены. Гидролиз эпимерной смеси аддуктов хлорнитрила позволил получить желаемый бициклогептанон с высоким выходом.

Сэмюэл Дж. Данишефски использовал реакцию Дильса-Альдера для синтеза префената динатрия, биосинтетического предшественника аминокислот фенилаланина и тирозина, в 1979 году. Эта последовательность примечательна как одна из самых ранних включить в общий синтез 1-метокси-3-силоксибутадиен, так называемый диен Данишефского. Его полезность будет очевидна ниже, а именно, для получения α, β-ненасыщенных циклогексеноновых систем.
. В своем синтезе резерпина в 1980 году Пол Вендер и его коллеги использовали реакцию Дильса-Альдера, чтобы установить цис-декалиновый каркас D и E колец природного продукта. Первоначальная схема Дильса-Альдера между 2-ацетоксиакриловой кислотой и 1,2-дигидропиридин-1-карбоксилатом, показанная ниже, ставит вновь установленную карбоксильную группу в положение для перегруппировки исключительно в цис-конденсированные кольца после преобразования в изохинуклиден, показанный ниже. Цис-слияние позволило установить стереохимию у C17 и C18: сначала путем отщепления ацетатной группы у C18 с образованием кетона, который может модулировать стереохимию метоксигруппы C17, а затем путем восстановления кетона у C18 из экзо-лицо для достижения стереохимии конечного продукта.

В Стивене Ф. Мартине синтезе резерпина цис-конденсированные кольца D и E также были образованы реакцией Дильса-Альдера. Внутримолекулярное соединение пиранона по Дильсу-Альдеру ниже с последующей экструзией диоксида углерода через ретро [4 + 2] дает бициклический лактам. Эпоксидирование с менее затрудненной α-грани с последующим раскрытием эпоксида на менее затрудненной C18 дало желаемую стереохимию в этих положениях, в то время как цис-синтез достигался с помощью гидрогенизации, опять же, в основном, с менее затрудненной стороны.

Пиранон также использовался в качестве диенофила К. Группа С. Николау в тотальном синтезе таксола. Межмолекулярная реакция гидроксипирона и α, β-ненасыщенного сложного эфира, показанная ниже, страдает низким выходом и региоселективностью; однако под действием фенилбороновой кислоты желаемый аддукт может быть получен с выходом 61% после расщепления бороната 2,2-диметил-1,3-пропандиолом. Стереоспецифичность реакции Дильса-Альдера в этом случае позволила определить четыре стереоцентра, которые были перенесены в конечный продукт.

Реакция Дильса-Альдера была ключевым этапом в синтезе (-) - фурахиноцина C Амосом Смитом. Дион 1 был преобразован в необходимый диен путем енолизации с использованием двух последовательных силилирований с TMSCl. За циклоприсоединением Дильса-Альдера с бромхиноном следовало спонтанное дегидрогалогенирование с повторным образованием ароматического кольца. Диен в этом случае примечателен как редкий пример циклического производного диена Данишефского.

и в своем синтезе таберсонина в 1998 году использовали метод Дильса-Альдера для установления цис-относительной стереохимии алкалоидного ядра. Превращение цис-альдегида в соответствующий ему алкен олефинированием по Виттигу и последующим метатезисом с замыканием цикла с катализатором Шрока дало второе кольцо алкалоидного ядра. Диен в этом случае примечателен как пример 1-амино-3-силоксибутадиена, иначе известного как диен Равала.

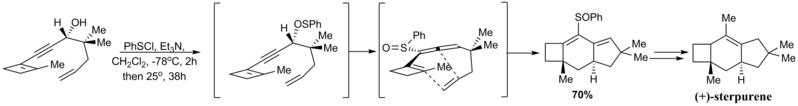
В 1988 г. и Ричард Гиббс сообщил об энантиоселективном синтезе (+) - стерпурена, который показал замечательную внутримолекулярную реакцию Дильса-Альдера аллена. [2,3] -сигматропная перегруппировка тиофенильной группы с образованием сульфоксида, как показано ниже, протекала энантиоспецифично из-за предварительно определенной стереохимии пропаргилового спирта. Таким образом, образующийся единичный изомер аллена может привести к появлению диена-диена только на одной стороне образовавшегося «диена».
Синтез (-) - тетрациклина Эндрю Майерсом в 2005 г. позволил получить линейное тетрациклическое ядро антибиотика с помощью реакции Дильса-Альдера. При термическом инициировании одновременного открытия бензоциклобутена образуется о-хинодиметан, который взаимодействует межмолекулярно с образованием тетрациклинового скелета; показанный диастереомер затем кристаллизовали из метанола после очистки колоночной хроматографией. Авторы отмечают, что свободная гидроксильная группа диенофила была неотъемлемой частью успеха реакции, поскольку гидроксил-защищенные варианты не реагировали в нескольких различных условиях реакции.

Такемура и др. синтезировал кантарадрин в 1980 году Дильсом-Альдером с использованием высокого давления.
Синтетические применения реакции Дильса-Альдера были подробно рассмотрены.