
Войти
 Расположение MT-CO2 ge ne в митохондриальном геноме человека. MT-CO2 является одним из трех митохондриальных генов субъединицы цитохром с оксидазы (оранжевые прямоугольники).
Расположение MT-CO2 ge ne в митохондриальном геноме человека. MT-CO2 является одним из трех митохондриальных генов субъединицы цитохром с оксидазы (оранжевые прямоугольники). | Субъединица цитохром с оксидазы II, трансмембранный домен | |||||||||
|---|---|---|---|---|---|---|---|---|---|
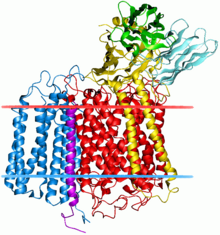 Бактериальный комплекс цитохром с оксидазы. Субблок II обозначен синим. Бактериальный комплекс цитохром с оксидазы. Субблок II обозначен синим. | |||||||||
| Идентификаторы | |||||||||
| Символ | COX2_TM | ||||||||
| Pfam | PF02790 | ||||||||
| InterPro | IPR011759 | ||||||||
| PROSITE | PDOC00075 | ||||||||
| SCOPe | 1occ / SUPFAM | ||||||||
| TCDB | 3.D.4 | ||||||||
| суперсемейство OPM | 4 | ||||||||
| белок OPM | 1v55 | ||||||||
| |||||||||
| Субъединица II цитохром С оксидазы, периплазматический домен | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Идентификаторы | |||||||||
| Символ | COX2 | ||||||||
| Pfam | PF00116 | ||||||||
| InterPro | IPR002429 | ||||||||
| CDD | cd13912 | ||||||||
| |||||||||
Субъединица 2 цитохром с оксидазы, также известная как полипептид цитохром с оксидазы II, представляет собой белок, который у человека кодируется геном MT-CO2 . Субъединица II цитохром с оксидазы, сокращенно COXII, COX2, COII или MT-CO2, является второй субъединицей цитохром с оксидаза. Он также является одной из трех субъединиц митохондриальной ДНК (мтДНК) (MT-CO1, MT-CO2, MT-CO3 ) респираторного комплекс IV.
Ген MT-CO2 расположен на p плече митохондриальной ДНК в положении 12 и охватывает 683 пары оснований. Ген MT-CO2 продуцирует белок 25,6 кДа, состоящий из 227 аминокислот. MT-CO2 является субъединицей фермента цитохром с оксидазы (EC 1.9.3.1 ) (Комплекс IV), олигомерного ферментативного комплекса митохондриальной дыхательной цепи участвует в переносе электронов от цитохрома c к кислороду. У эукариот этот ферментный комплекс расположен во внутренней мембране митохондрий ; у аэробных прокариот он обнаружен в плазматической мембране. Ферментный комплекс состоит из 3-4 субъединиц (прокариот ) до 13 полипептидов (млекопитающие). N-концевой домен цитохром С оксидазы содержит две трансмембранные альфа-спирали. Как известно, структура MT-CO2 содержит один окислительно-восстановительный центр и биядерный медный A-центр (CuA). CuA располагается в консервативной цистеиновой петле в положениях 196 и 200 аминокислот и консервативном гистидине в положениях 204. Некоторые бактериальные MT- CO2 имеет С-концевое удлинение, которое содержит ковалентно связанный гем c.
Ген MT-CO2 кодирует вторую субъединицу цитохром с оксидазы (комплекс IV), компонент митохондриальной дыхательной цепи, который катализирует восстановление кислорода до воды. MT-CO2 является одной из трех субъединиц, которые ответственны за формирование функционального ядра цитохром с оксидазы. MT-CO2 играет важную роль в переносе электронов от цитохрома c к биметаллическому центру каталитической субъединицы 1 за счет использования его биядерного медного A-центра. Он содержит две смежные трансмембранные области на своем N-конце, и большая часть белка экспонируется в периплазматическое или митохондриальное межмембранное пространство соответственно. MT-CO2 обеспечивает сайт связывания субстрата и содержит биядерный медный A-центр, вероятно, первичный акцептор цитохром-соксидазы.
Варианты MT-CO2 были связаны с дефицитом митохондрий Комплекса IV, дефицитом ферментного комплекса митохондриальной дыхательной цепи который катализирует окисление цитохрома c с использованием молекулярного кислорода. Дефицит характеризуется гетерогенными фенотипами в диапазоне от изолированной миопатии до тяжелого мультисистемного заболевания, поражающего несколько тканей и органов. К другим клиническим проявлениям относятся гипертрофическая кардиомиопатия, гепатомегалия и нарушение функции печени, гипотония, мышечная слабость, непереносимость упражнений, задержка развития, задержка двигательного развития и умственная отсталость. Известно также, что мутации MT-CO2 вызывают болезнь Ли, которая может быть вызвана аномалией или дефицитом цитохромоксидазы.
. У пациентов с патогенными мутациями обнаружен широкий спектр симптомов. в гене MT-CO2 с дефицитом митохонридов Комплекса IV. Обнаружено, что делеционная мутация одного нуклеотида (7630delT) в гене вызывает симптомы обратимой афазии, правого гемипареза, гемианопсии, непереносимость физических упражнений, прогрессирующее умственное нарушение и низкий рост. Кроме того, у пациента с бессмысленной мутацией (7896G>A) гена наблюдались такие фенотипы, как низкий рост, низкий вес, микроцефалия, кожные аномалии, тяжелая гипотония и нормальные рефлексы. Новая гетероплазматическая мутация (7587T>C), которая изменила кодон инициации гена MT-CO2 у пациентов, продемонстрировала такие клинические проявления, как прогрессирующая походка атаксия, когнитивные нарушения, двусторонняя атрофия зрительного нерва, пигментная ретинопатия, снижение цветового зрения и легкое дистальное истощение мышц.
Ювенильная миопатия, энцефалопатия, лактоацидоз и инсульт также были связаны с мутациями в гене MT-CO2.
Известно, что MT-CO2 взаимодействует с цитохромом c за счет использования лизинового кольца вокруг карбоксила, содержащего гем. край из цитохрома c в MT-CO2, включая глутамат 129, аспартат 132 и глутамат 19.
. Эта статья включает текст из Национальная медицинская библиотека США, которая находится в общественном достоянии.
