
Войти
 Технологии захвата конформации хромосомы
Технологии захвата конформации хромосомы Методы захвата конформации хромосомы (часто сокращенно называют технологиями 3C или методами на основе 3C) представляют собой набор методов молекулярной биологии, используемых для анализа пространственной организации хроматина в клетке. Эти методы количественно определяют количество взаимодействий между геномными локусами, которые находятся рядом в трехмерном пространстве, но могут быть разделены множеством нуклеотидов в линейном геноме. Такие взаимодействия могут быть результатом биологических функций, таких как взаимодействия промотор - энхансер, или случайного образования петель полимера, когда ненаправленное физическое движение хроматина вызывает столкновение локусов. Частоты взаимодействия могут быть проанализированы напрямую, или они могут быть преобразованы в расстояния и использованы для реконструкции трехмерных структур.
Основное различие между методами, основанными на 3C, заключается в их объеме. Например, при использовании ПЦР для обнаружения взаимодействия в эксперименте 3С взаимодействия между двумя конкретными фрагментами оцениваются количественно. Напротив, Hi-C количественно оценивает взаимодействия между всеми возможными парами фрагментов одновременно. Глубокое секвенирование материала, производимого 3C, также дает карты взаимодействий в масштабе всего генома.
Исторически микроскопия был основным методом исследования ядерной организации, который можно датировать 1590 годом.

Все методы 3C начинаются с аналогичного набора шагов, выполняемых на выборке ячеек.
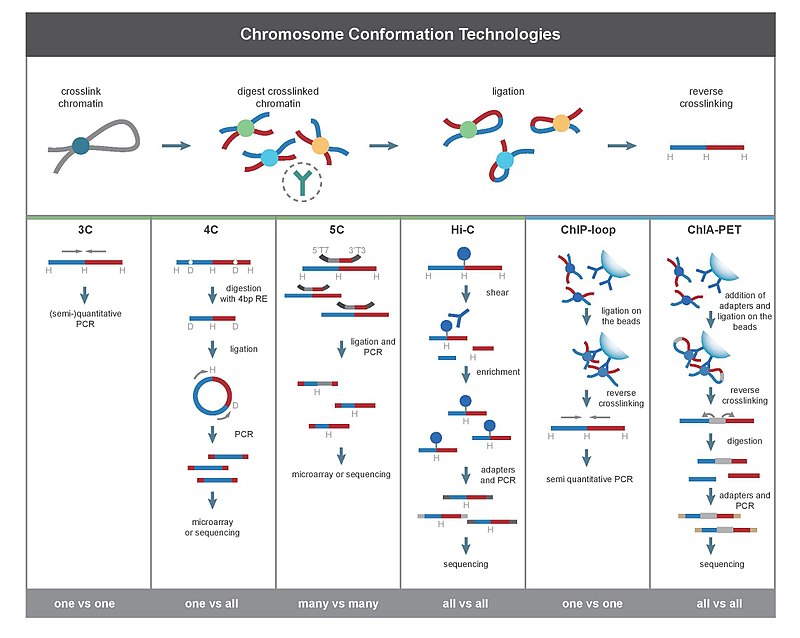
Во-первых, геномы клеток сшиты с формальдегидом, который вводит связи, которые «замораживают» взаимодействия между геномными локусами. Обработка клеток 1-3% формальдегидом в течение 10-30 минут при комнатной температуре является наиболее распространенной, однако необходима стандартизация для предотвращения высокого перекрестного связывания белка с ДНК, поскольку это может отрицательно повлиять на эффективность рестрикционного переваривания на последующем этапе. Затем геном разрезают на фрагменты с помощью эндонуклеазы рестрикции. Размер ограничительных фрагментов определяет разрешение отображения взаимодействий. Для этой цели используются рестрикционные ферменты (RE), которые разрезают последовательности распознавания 6 п.н., такие как EcoR1 или HindIII, поскольку они разрезают геном каждые 4000 п.н., что дает ~ 1 миллион фрагменты в геноме человека. Для более точного отображения взаимодействий также может использоваться RE, распознающий 4bp. Следующим шагом является лигирование на основе близости. Это происходит при низких концентрациях ДНК или внутри интактных, проницаемых ядер в присутствии ДНК-лигазы Т4, так что лигирование между перекрестно-сшитыми взаимодействующими фрагментами предпочтительнее лигирования между фрагментами, которые не перекрестно связаны. Затем взаимодействующие локусы количественно оцениваются путем амплификации лигированных соединений методами ПЦР.
Эксперимент по захвату конформации хромосомы (3C) количественно определяет взаимодействия между одной парой геномных локусов. Например, 3C можно использовать для тестирования взаимодействия кандидата-промотора-энхансера. Лигированные фрагменты выявляют с помощью ПЦР с известными праймерами. Вот почему этот метод требует предварительного знания взаимодействующих областей.
Захват конформации хромосомы на чипе (4C) фиксирует взаимодействия между одним локусом и всеми другими геномными локусами. Он включает в себя вторую стадию лигирования, чтобы создать фрагменты ДНК с самоциклической циркуляцией, которые используются для выполнения обратной ПЦР. Обратная ПЦР позволяет использовать известную последовательность для амплификации присоединенной к ней неизвестной последовательности. В отличие от 3C и 5C, метод 4C не требует предварительного знания обоих взаимодействующих хромосомных областей. Результаты, полученные с помощью 4C, хорошо воспроизводимы, при этом большинство взаимодействий обнаруживаются между областями, расположенными рядом друг с другом. На одном микрочипе можно проанализировать примерно миллион взаимодействий.
Копия захвата конформации хромосомы (5C) обнаруживает взаимодействия между всеми рестрикционными фрагментами в пределах данного регион, размер которого обычно не превышает мегабазы. Это делается путем лигирования универсальных праймеров ко всем фрагментам. Однако 5C имеет относительно низкое покрытие. Метод 5C преодолевает проблемы соединений на этапе внутримолекулярного лигирования и полезен для построения сложных взаимодействий конкретных интересующих локусов. Этот подход не подходит для проведения сложных взаимодействий в масштабе всего генома, поскольку для этого потребуются миллионы праймеров 5C.
Hi-C использует высокопроизводительное секвенирование для поиска нуклеотидной последовательности фрагментов и использует секвенирование парных концов, которое извлекает короткие последовательности с каждого конца каждого лигированного фрагмента. Таким образом, для данного лигированного фрагмента две полученные последовательности должны представлять два разных рестрикционных фрагмента, которые были лигированы вместе на стадии лигирования на основе близости. Пара последовательностей индивидуально выравнивается по геному, таким образом определяя фрагменты, участвующие в этом событии лигирования. Следовательно, проверяются все возможные парные взаимодействия между фрагментами.
В ряде методов используется захват олигонуклеотидов для обогащения библиотек 3C и Hi-C для конкретных интересующих локусов. Эти методы включают Capture-C, NG Capture-C, Capture-3C и Capture Hi-C. Эти методы могут обеспечить более высокое разрешение и чувствительность, чем методы на основе 4C.
Одноячеечные адаптации этих методов, такие как ChIP-seq и Hi-C, могут быть используется для исследования взаимодействий, происходящих в отдельных клетках.
ChIP-loop объединяет 3C с ChIP-seq для обнаружения взаимодействий между двумя интересующими локусами, опосредованными интересующим белком. ChIP-петля может быть полезна для идентификации дальнодействующих цис-взаимодействий и транс-взаимодействий, опосредованных белками, поскольку частые столкновения ДНК не происходят.
ChIA-PET объединяет Hi- C с ChIP-seq для обнаружения всех взаимодействий, опосредованных интересующим белком. HiChIP был разработан для проведения анализа, аналогичного ChIA-PET, с меньшим количеством входящего материала.
Методы 3C привели к ряду биологических открытий, включая открытие новых структурных особенностей хромосом, каталогизация петель хроматина и лучшее понимание механизмов регуляции транскрипции (нарушение которых может привести к заболеванию).
Методы 3C продемонстрировали важность пространственной близости регуляторных элементов к гены, которые они регулируют. Например, в тканях, которые экспрессируют гены глобина, контролирующая область локуса β-глобина образует петлю с этими генами. Эта петля не обнаруживается в тканях, где ген не экспрессируется. Эта технология также способствовала генетическому и эпигенетическому изучению хромосом как у модельных организмов, так и у людей.
Эти методы выявили крупномасштабную организацию генома в топологически связанные домены (TAD), которые коррелируют с эпигенетическими маркерами. Некоторые TAD транскрипционно активны, а другие репрессированы. Многие TAD были обнаружены у D. melanogaster, мыши и человека. Более того, CTCF и когезин играют важную роль в определении TAD и взаимодействий энхансер-промотор. Результат показывает, что ориентация связывающих мотивов CTCF в петле энхансер-промотор должна быть обращена друг к другу, чтобы энхансер нашел свою правильную мишень.
Есть несколько заболевания, вызванные дефектами взаимодействий промотор-энхансер, который рассматривается в этой статье.
Бета-талассемия - это определенный тип заболеваний крови, вызванных делецией элемента энхансера LCR.
Голопрозэнцефалия является цефалической. расстройство, вызванное мутацией в элементе энхансера SBE2, которая, в свою очередь, ослабляет продукцию гена SHH.
PPD2 (полидактилия трехфалангового пальца) вызывается мутацией энхансера ZRS, которая, в свою очередь, усиливает продукцию гена SHH.
Аденокарцинома легкого может быть вызвана дупликацией энхансерного элемента гена MYC.
Т-клеточный острый лимфобластный лейкоз вызван введением нового энхансера.
 Тепловая карта и круговая визуализация данных Hi-C. а. Взаимодействия Hi-C между всеми хромосомами из клеток почек человека G401, как показано программой my5C. б. Визуализация тепловой карты, иллюстрирующая двудольную структуру X-хромосомы мыши, построенная Hi-Browse. c. Визуализация тепловой карты локуса 3 Мбит / с (chr4: 18000000-21000000), произведенная Juicebox, с использованием данных Hi-C in-situ из линии клеток GM12878. d. Круговой график двудольной X-хромосомы мыши, созданный браузером эпигенома. Изображение из
Тепловая карта и круговая визуализация данных Hi-C. а. Взаимодействия Hi-C между всеми хромосомами из клеток почек человека G401, как показано программой my5C. б. Визуализация тепловой карты, иллюстрирующая двудольную структуру X-хромосомы мыши, построенная Hi-Browse. c. Визуализация тепловой карты локуса 3 Мбит / с (chr4: 18000000-21000000), произведенная Juicebox, с использованием данных Hi-C in-situ из линии клеток GM12878. d. Круговой график двудольной X-хромосомы мыши, созданный браузером эпигенома. Изображение из Различные эксперименты в стиле 3C дают данные с очень разными структурами и статистическими свойствами. Таким образом, для каждого типа экспериментов существуют определенные пакеты анализа.
Данные Hi-C часто используются для анализа организации хроматина в масштабе всего генома, например, топологически ассоциированные домены (TAD), линейно смежные области генома, связанные в трехмерном пространстве. Было разработано несколько алгоритмов для идентификации TAD по данным Hi-C.
Hi-C и его последующий анализ развиваются. Fit-Hi-C - это метод, основанный на подходе дискретного бинирования с модификациями добавления расстояния взаимодействия (начальная подгонка сплайна, также известного как сплайн-1) и уточнения нулевой модели (сплайн-2). Результатом Fit-Hi-C является список попарных внутрихромосомных взаимодействий с их p-значениями и q-значениями.
Трехмерная организация генома также может быть проанализирована с помощью eigendecomposition контактной матрицы. Каждый собственный вектор соответствует набору локусов, которые не обязательно линейно смежны, но имеют общие структурные особенности.
Существенным мешающим фактором в технологиях 3C являются частые неспецифические взаимодействия между геномными локусами, возникающие из-за случайных поведение полимера. Взаимодействие между двумя локусами должно быть подтверждено как специфическое посредством тестирования статистической значимости.
Существует два основных способа нормализации исходных тепловых карт контактов Hi-C. Первый способ - предположить равную видимость, то есть существует равный шанс взаимодействия каждой хромосомной позиции. Следовательно, истинный сигнал карты контактов Hi-C должен быть сбалансированной матрицей (сбалансированная матрица имеет постоянные суммы строк и суммы столбцов). Примером алгоритмов, предполагающих равную видимость, является алгоритм Синкхорна-Кноппа, который масштабирует необработанную карту контактов Hi-C в сбалансированную матрицу.
Другой способ - предположить, что с каждым положением хромосомы связано смещение. Значение карты контакта в каждой координате будет истинным сигналом в этой позиции, умноженной на смещение, связанное с двумя позициями контакта. Примером алгоритмов, направленных на решение этой модели смещения, является итеративная коррекция, которая итеративно регрессировала смещение строк и столбцов из необработанной карты контактов Hi-C. Существует ряд программных инструментов, доступных для анализа данных Hi-C.
Мотивы ДНК - это конкретные короткие последовательности ДНК, часто длиной 8-20 нуклеотидов, которые статистически чрезмерно представлены в наборе последовательностей с общей биологической функцией. В настоящее время регуляторные мотивы дальнодействующих взаимодействий хроматина недостаточно изучены. Несколько исследований были сосредоточены на выяснении влияния мотивов ДНК на взаимодействия промотор-энхансер.
Бейли и др. идентифицировал, что мотив ZNF143 в промоторных областях обеспечивает специфичность последовательности для взаимодействий промотор-энхансер. Мутация мотива ZNF143 снизила частоту взаимодействий промотор-энхансер, что свидетельствует о том, что ZNF143 является новым фактором образования петель хроматина.
Для анализа мотивов в масштабе генома в 2016 г. Wong et al. сообщили о списке из 19 491 пары мотивов ДНК для линии клеток K562 о взаимодействиях промотор-энхансер. В результате они предположили, что множественность спаривания мотивов (количество мотивов, спаренных с данным мотивом) связана с расстоянием взаимодействия и типом регуляторной области. В следующем году Вонг опубликовал еще одну статью, в которой сообщалось о 18 879 парах мотивов в 6 линиях клеток человека. Новым вкладом этой работы является MotifHyades, инструмент для обнаружения мотивов , который можно напрямую применять к парным последовательностям.
Методы, основанные на 3C, могут дать представление о хромосомных перестройках в геномах рака. Более того, они могут показать изменения пространственной близости регуляторных элементов и их генов-мишеней, что дает более глубокое понимание структурной и функциональной основы генома.