
Войти
Эпигенетика рака - это исследование эпигенетических модификаций ДНК раковых клеток, которые не связаны изменением нуклеотидной системы, а вместо этого изменяет способ изменения генетического кода. Эпигенетические изменения могут быть столь же важны или даже более важны, чем генетические мутации в трансформации клетки в рак. При раке потеря экспрессии генов происходит примерно в 10 раз чаще из-за подавления транскрипции (вызванного гиперметилированием эпигенетического промотора CpG-островков ), чем из-за мутаций. Как Vogelstein et al. Отметим, что при колоректальном раке обычно бывает от 3 до 6 мутаций водителя и от 33 до 66 автостопщиков или мутаций пассажира. Однако волях толстой кишки по соседней слизистой оболочке, которая выглядит нормально, в промоторах генов опухолей имеется от 600 до 800 сильно метилированных CpG-островков, в то время как эти CpG-островки не метилированы в соседней слизистой оболочке. Манипуляции с эпигенетическими изменениями открывают большие перспективы для профилактики, лечения и лечения рака. При различных типах рака могут быть нарушены различные эпигенетические механизмы, такие как подавление генов-супрессоров опухолей и активация онкогенов измененным CpG островковые паттерны метилирования, модификации гистонов и нарушение регуляции ДНК-связывающих белков. Несколько лекарств, имеющего эпигенетическое действие, теперь используются при некоторых из этих заболеваний.
 Паттерны эпигенетики в нормальных и раковых клетках
Паттерны эпигенетики в нормальных и раковых клетках  Эпигенетические изменения в прогрессии опухоли
Эпигенетические изменения в прогрессии опухоли  A ДНК фрагмент молекулы, который метилирован по двум цитозинам
A ДНК фрагмент молекулы, который метилирован по двум цитозинам В соматических клетках паттерны метилирования ДНК обычно передаются дочерними клетками с высокой точностью. Обычно это метилирование происходит только в цитозинах, которые расположены 5 'от гуанозина в динуклеотидах CpG эукариот более высокого порядка. Однако эпигенетическим метилирование ДНК различается между нормальными клетками и опухолевыми клетками человека. «Нормальный» профиль метилирования CpG часто инвертируется в клетках, которые становятся онкогенными. В нормальных клетках островки CpG, предшествующие промоторам гена, обычно и имеют тенденцию быть транскрипционно активными, в то время как другие отдельные динуклеотиды CpG по всему геному имеют тенденцию метилироваться.. Однако в раковых клетках CpG-островки, предшествующие промоторам гена-супрессора опухоли, часто гиперметилированы, в то время как CpG-метилирование промоторных областей онкогена и паразитарных повторяющихся последовательностей часто снижено.
Гиперметилирование промоторных органов гена-супрессора может привести к подавлению этих генов. Этот тип эпигенетической мутации позволяет клеткам бесконтрольно расти и воспроизводиться, что приводит к онкогенезу. Добавление вокруг метильных групп к цитозинам приводит к тому, что ДНК плотно обвивается гистоновых белков, в результате чего ДНК не может подвергаться транскрипции (транскрипционно замалчивающая ДНК). Гены, обычно подавляемые транскрипцией из-за гиперметилирования промотора, включают: ингибитор циклин-зависимой киназы p16, ингибитор клеточного цикла; MGMT, ген репарации ДНК ; APC, регулятор клеточного цикла; MLH1, ген репарации ДНК; и BRCA1, другой ген репарации ДНК. Действительно, раковые клетки могут стать зависимыми от транскрипционного сайленсинга из-за гиперметилирования промоторов некоторых генов-супрессоров опухолей, процесса, известного как эпигенетическая зависимость.
Гипометилирование динуклеотидов CpG в других частях генома приводит к нестабильность хромосомы из-за таких механизмов, как потеря импринтинга и реактивация мобильных элементов. Потеря импринтинга гена инсулиноподобного фактора роста (IGF2) увеличивает риск колоректального рака и связана с синдромом Беквита-Видеманна, что значительно увеличивает риск рака новорожденных. В здоровых клетках динуклеотиды CpG с более низкой плотностью обнаруживаются в кодирующих и некодирующих межгенных областях. Экспрессия некоторых повторяющихся последовательностей и мейотическая рекомбинация в центрере подавляются посредством метилирования
Весь геном раковой клетки содержит значительно меньше метилцитозина, чем геном здоровой клетки. Фактически, геномы раковых клеток на 20-50% меньше метилирования отдельных динуклеотидов CpG по всему геному. Островки CpG, обнаруженные в промоторных областях, обычно защищены от метилирования ДНК. В раковых клетках CpG-островки гипометилированы. Области, прилегающие к CpG-островам, называемые берегами CpG-островков, место, где большая часть метилирования ДНК в контексте динуклеотидов CpG. Раковые клетки дифференцированно метилированы на берегах острова CpG. В раковых клетках гиперметилирование на берегах CpG-островков перемещается на CpG-острова, или гипометилирование CpG-островков перемещается на берега CpG-островов, устраняя резкие эпигенетические границы между этими генетическими элементами. В раковых клетках «глобальное гипометилирование» из-за нарушения ДНК-метилтрансфераз (DNMT) может вызвать организм митотической рекомбинации и хромосомной перестройке, что в итоге приводит к анеуплоидии когда хромосомы не могут должным образом разделиться во время митоза.
Метилирование CpG-островков важно для регуляции экспрессии генов, однако метилирование цитозина может напрямую вести к дестабилизирующим генетическим мутациям и предраковому состоянию клеток. Метилированные цитозины делают гидролиз аминогруппы и спонтанное превращение в тимин более благоприятными. Они могут вызывать аберрантный набор белков хроматина. Метилирование цитозина изменяет структуру УФ-света нуклеотидным основанием, создавая димеры пиримидина. Когда мутация приводит к потере гетерозиготности на сайтах гена-супрессора опухоли, эти гены могут стать неактивными. Мутации одной пары оснований во время репликации также могут иметь пагубные последствия.
Эукариотическая ДНК имеет сложную структуру. Обычно он обернут вокруг специальных белков, называемых гистонами, чтобы сформировать структуру, называемую нуклеосомой. Нуклеосома состоит из 2 наборов по 4 гистона: H2A, H2B, H3 и H4. Кроме того, гистон H1 обеспечивает ДНК вне нуклеосомы. Определенные ферменты, модифицирующие гистоны, функции или удаление функциональных групп гистонов, и эти модификации влияют на уровень транскрипции генов, обнутых вокруг этих гистонов, и на уровень репликации ДНК. Профили модификации гистонов здоровых и раковых клеток начало различаться.
По сравнению со здоровыми клетками, раковые клетки демонстрируют пониженное содержание моноацетилированных и триметилированных форм гистона H4 (пониженное содержание H4ac и H4me3). Кроме того, мышиные модели показали, что уменьшение асимметричного диметилирования гистона H4R3 (H4R3me2a) промотора p19ARF коррелирует с более поздними случаями онкогенеза и метастазирования. В моделях на мышах потеря ацетилирования и триметилирования гистона H4 увеличивается по мере продолжения роста опухоли. Потеря ацетилирования гистона H4 Лизин 16 (H4K16ac ), который является признаком старения на теломерах, в частности, теряет ацетилирование. Некоторые ученые надеются, что с помощью этой специальной потерей ацетилирования гистонов можно бороться с помощью ингибитора гистондеацетилазы (HDAC), специфичного для SIRT1, HDAC, специфичного для H4K16.
Другой гистон Признаки, связанный с туморогенезом, включает повышенное деацетилирование (снижение ацетилирования) гистонов H3 и H4, пониженное гистон H3, лизин 4 (H3K4me3 ) и повышенное монометилирование гистона H3, лизин 9 () и триметилирование гистона H3, лизина 27 (H3K27me3 ). Эти модификации гистонов могут заглушить-супрессоры опухоли, несмотря на снижение метилирования CpG-островка гена (событие, которое обычно активирует гены).
Некоторые исследования были сосредоточены на блокировании действия BRD4 на ацетилированные гистоны, которые, как было показано, увеличивают экспрессию белка Myc, участвуют в нескольких рака. Процесс разработки препарата для связывания с BRD4 примечателен совместным открытым подходом, который использует команда.
Ген супрессора опухолей p53 ет репарацию ДНК и может вызвать апоптоз в клетках с нарушенной регуляцией. E Soto-Reyes и F. Recillas-Targa показали важность белка CTCF в регуляции экспрессии p53. CTCF, или фактор связывания CCCTC, представляет собой белок цинковый палец, который изолирует промотор p53 от накопления репрессивных гистоновых меток. В некоторых типах раковых клеток белок CTCF не связывается нормально, и промотор p53 накапливает репрессивные гистоновые метки, вызывая снижение экспрессии p53.
Также могут возникать мутации в самом эпигенетическом механизме, первые ответственные за изменяющиеся эпигенетические профили раковых клеток. Варианты гистона семейства H2A высокоативны у млекопитающих, обнаруживающие критическую роль в регуляции многих ядерных процессов, изменяя структуру хроматина. Один из вариантов вариантов H2A, H2A.X отмечает повреждение ДНК, облегчение набора белков репарации ДНК для восстановления целостности генома. Другой вариант, H2A.Z, играет важную роль в активации, так и в репрессии генов. Высокий уровень экспрессии H2A.Z имеет место при высокой степени степени с клеточной пролиферацией и геномной нестабильностью. Вариант гистона macroH2A1 важен в патогенезе многих типов рака, например, гепатоцеллюлярной карциномы. Другие механизмы включают снижение активности H4K16ac, которое может быть вызвано либо снижением активности гистонацетилтрансферазы (HATs), либо деацетилирования с помощью SIRT1. Аналогичным образом, инактивирующая мутация сдвига рамки считывания в HDAC2, гистондеацетилаза, которая действует на многие лизины гистонового хвоста , была связана с раком. демонстрирует измененные паттерны ацетилирования гистонов. Эти открытия указывают на многообещающий механизм изменения эпигенетических профилей посредством ферментативного ингибирования или усиления.
Повреждение ДНК, вызванное ультрафиолетовым светом, ионизирующим излучением, токсическими веществами окружающей среды и экологическими химическими веществами, также может привести к нестабильности генома и раку. Реакция на повреждение ДНКцепочечной (DSB) частично опосредуется модификациями гистонов. В DSB белковый комплекс MRE11 - RAD50 - NBS1 (MRN) рекрутирует атаксию телеангиэктазии мутантную (ATM) киназу, которая фосфорилирует серин 129 гистона 2А. MDC1, медиатор контрольной точки 1 повреждения ДНК, связывается с фосфопептидом, фосфорилированием H2AX может распространяться посредством петли положительной обратной связи рекрутирования и фосфорилирования MRN-ATM. TIP60 ацетилирует соединение, которое затем является., субъединица модуля восприимчивости к раку молочной железы типа 1 (BRCA1 -A) репарации ДНК, связывает убиквитин, прикрепленный к гистонам. Активность BRCA1-A останавливает клеточный цикл в контрольной точке G2 / M, давая время для восстановления ДНК, или может быть инициирован апоптоз.
У млекопитающих микроРНК (миРНК) регулируют около 60% транскрипционной активности генов, кодирующих белок. Некоторые miRNA также подвергаются замалчиванию, связанному с метилированием, в раковых клетках. Let-7 и miR15 / 16 играют важную роль в подавлении онкогенов RAS и BCL2 , и их подавление происходит в раковых клетках. Снижение экспрессии miR-125b1, miRNA, которая функционирует как опухолевый супрессор, наблюдали при раке простаты, яичников, груди и глиальных клеток. Эксперименты in vitro показали, что miR-125b1 нацелен на два гена, HER2 / neu и ESR1, которые связаны с раком груди. Метилирование ДНК, особенно гиперметилирование, является одним из способов эпигенетического подавления miR-125b1. У пациентов с раком груди наблюдалось гиперметилирование CpG-островков, наблюдалось проксимальнее сайта начала транскрипции. Утрата связывания CTCF и увеличение репрессивных гистоновых меток, H3K9me3 и H3K27me3, коррелируют с метилированием ДНК и подавлением miR-125b1. Механически CTCF может функционировать как пограничный элемент, чтобы распространение метилирования ДНК. Результаты экспериментов, проведенных Soto-Reyes et al. указывают на отрицательный эффект метилирования на функцию и экспрессию miR-125b1. Таким образом, они пришли к выводу, что метилирование ДНК участвует в подавлении гена. Кроме того, некоторые miRNA могут быть полезны в качестве опухолевых маркеров. Эпигенетическое подавление генов miRNA за счет аберрантного метилирования ДНК - частое явление в раковых клетках; почти треть промоторов miRNA, активных в нормальных клетках молочной железы, обнаружена гиперметилированная в клетках рака молочной железы - это в раз больше, чем обычно наблюдается для генов, кодирующих белок.
Нарушение регуляции метаболизма позволяет опухолевым клеткам генерировать необходимые строительные блоки, а также модулировать эпигенетические метки для поддержки инициации и прогрессирования рака. Вызванные раком метаболические изменения изменяют эпигенетический ландшафт, новые модификации гистонов и ДНК, тем самым способствуя злокачественной трансформации, адаптации к неадекватному питанию и метастазированию. Накопление метаболитов при раке может воздействовать на эпигенетические ферменты, чтобы глобально изменить эпигенетический ландшафт. Связанные с раком метаболические изменения приводят к локус-специфическому перекодированию эпигенетических меток. Эпигенетика рака может быть воспроизведена посредством 1) дозозависимой модуляции эпигенетики рака с помощью метаболитов; 2) специфичный для набора метаболических ферментов; и 3) нацеливание на эпигенетические ферменты с помощью сигналов питания.
Повреждение ДНК, по-видимому, является основной причиной основной причины рака. Если репарация ДНК недостаточна, повреждения ДНК тенденции к накоплению. Такое избыточное повреждение ДНК может увеличивать мутационные ошибки во время репликации ДНК из-за подверженного ошибкам транслезионного синтеза. Избыточное повреждение ДНК также может увеличивать эпигенетические изменения из-за ошибок во время восстановления ДНК. Такие мутации и эпигенетические изменения могут вызвать рак (см. злокачественные новообразования ).
Мутации зародышевой линии в генах репарации ДНК вызывают только 2–5% случаев рака толстой кишки. Причина, вызывающая дефицит репарации ДНК, часто используется причинным фактором для этих видов рака.
Сверхэкспрессия определенных miRNA может напрямую снижать экспрессию определенных репараций ДНК. Ван и др. относится к 6 генам репарации ДНК, которые напрямую нацелены на miRNA, образует в скобках: ATM (miR-421), RAD52 (miR-210, miR-373), RAD23B (miR-373), MSH2 (miR-21), BRCA1 (miR-182) и P53 (miR-504, miR-125b). Совсем недавно Tessitore et al. перечислили дополнительные гены репарации ДНК, напрямую нацеленные на дополнительные miRNA, включая ATM (miR-18a, miR-101), DNA-PK (miR-101), ATR (miR-185), Wip1 (miR-16), MLH1, MSH2 и MSH6 (miR-155), ERCC3 и ERCC4 (miR-192) и UNG2 (mir-16, miR-34c и miR-199a). Из этих miRNA, miR-16, miR-18a, miR-21, miR-34c, miR-125b, miR-101, miR-155, miR-182, miR-185 и miR-192 входят в число тех, которые были идентифицированы Шнекенбургером и Дидерих сверхэкспрессируется при раке толстой кишки за счет эпигенетического гипометилирования. Избыточная экспрессия любой из этих микроРНК может вызвать снижение экспрессии целевого гена репарации ДНК.
До 15% дефицита MLH1 при спорадическом раке толстой кишки, по-видимому, вызваны сверхэкспрессией микроРНК miR-155, которая подавляет экспрессию MLH1. Однако в большинстве из 68 спорадических случаев рака толстой кишки со сниженной экспрессией белка репарации ошибочного спаривания ДНК MLH1 обнаружен дефицит из-за эпигенетического метилирования острова CpG. гена MLH1.
В 28% глиобластом белок репарации ДНК MGMT недостаточен, но промотор MGMT не метилирован. В глиобластомах без метилированных промоторов MGMT уровень микроРНК miR-181d обратно коррелирует с экспрессией белка MGMT, а прямой мишенью miR-181d является MGMT мРНК 3'UTR (трехпраймовая нетранслируемая область мРНК MGMT). Таким образом, в 28% глиобластом повышенная экспрессия miR-181d и пониженная экспрессия фермента репарации ДНК MGMT могут быть причинным фактором. В 29–66% глиобластом репарация ДНК недостаточна из-за эпигенетического метилирования гена MGMT, что снижает экспрессию белка MGMT.
Белки группы A с высокой подвижностью (HMGA ), характеризующиеся АТ-крючком, представляют собой небольшие негистоновые ассоциированные с хроматином белки, которые могут модулировать транскрипцию. МикроРНК контролируют экспрессию белков HMGA, и эти белки (HMGA1 и HMGA2 ) являются элементами, контролирующими транскрипцию архитектурного хроматина. Palmieri et al. показали, что в нормальных тканях гены HGMA1 и HMGA2 нацелены (и, следовательно, сильно снижена в экспрессии) miR-15, miR-16, miR-26a, miR-196a2 и Let-7a.
Экспрессия HMGA почти не обнаруживается в дифференцированных тканях взрослого человека, но повышена при многих раках. Белки HGMA представляют собой полипептиды из ~ 100 аминокислотных остатков, характеризующиеся модульной организацией последовательностей. Эти белки имеют три высоко положительно заряженных области, называемых АТ-крючками, которые связывают малую бороздку участков ДНК, богатых АТ, в определенных областях ДНК. Неоплазии человека, включая рак щитовидной железы, предстательной железы, шейки матки, колоректального рака, поджелудочной железы и яичников, демонстрируют сильное увеличение белков HMGA1a и HMGA1b. У трансгенных мышей с HMGA1, нацеленным на лимфоидные клетки, развивается агрессивная лимфома, показывая, что высокая экспрессия HMGA1 не только связана с раком, но и что ген HMGA1 может действовать как онкоген, вызывая рак. Baldassarre et al. Показали, что белок HMGA1 связывается с промоторной областью гена репарации ДНК BRCA1 и ингибирует активность промотора BRCA1. Они также показали, что, хотя только 11% опухолей молочной железы имели гиперметилирование гена BRCA1, 82% агрессивных раковых опухолей молочной железы имеют низкую экспрессию белка BRCA1, и большинство этих сокращений былосвязано с ремоделированием хроматина высоких уровней белка HMGA1. Белок
HMGA2 специфически нацелен на промотор ERCC1, тем самым снижая экспрессию этого гена репарации ДНК. Экспрессия белка ERCC1 была недостаточной в 100% из 47 оцененных случаев рака толстой кишки (степень участия HGMA2 неизвестна).
Palmieri et al. показывает, что каждая из миРНК, нацеленных на гены HMGA, резко снижена почти во всех изученных аденомах гипофиза человека по сравнению с нормальным гипофизом. В соответствии с подавлением этих микроРНК, нацеленных на HMGA, наблюдалось увеличение количества мРНК, специфичных для HMGA1 и HMGA2. Три из этих микроРНК (miR-16, miR-196a и Let-7a) имеют метилированные промоторы и, следовательно, низкую экспрессию при раке толстой кишки. Для двух из них, miR-15 и miR-16, кодирующие области эпигенетически подавляются при раке из-за активности гистондеацетилазы. Когда эти микроРНК экспрессируются на низком уровне, белки HMGA1 и HMGA2 экспрессируются на высоком уровне. HMGA1 и HMGA2 нацелены (снижают экспрессию) генов репарации ДНК BRCA1 и ERCC1. Таким образом, репарация ДНК может быть уменьшена, что вероятно, обеспечение прогрессирования рака.
 Таблица распространенных повреждающих агентов ДНК, примеры повреждений, используемые для восстановления этих повреждений. Также показаны многие из генов этих путей, что указывает на то, какие гены эпигенетически регулируются, чтобы иметь сниженную (или повышенную) экспрессию при различных раковых заболеваниях. Он также показывает гены в пути соединения концов, подверженного ошибкам, опосредованному микрогомологией, с повышенной экспрессией при различных раковых заболеваниях.
Таблица распространенных повреждающих агентов ДНК, примеры повреждений, используемые для восстановления этих повреждений. Также показаны многие из генов этих путей, что указывает на то, какие гены эпигенетически регулируются, чтобы иметь сниженную (или повышенную) экспрессию при различных раковых заболеваниях. Он также показывает гены в пути соединения концов, подверженного ошибкам, опосредованному микрогомологией, с повышенной экспрессией при различных раковых заболеваниях. В этом разделе показаны некоторые часто встречающиеся агенты, повреждающие ДНК, примеры повреждений ДНК, которые вызывают их пути, борются с ними. Повреждения ДНК. По крайней мере, 169 ферментов практически используются в репарации ДНК, либо на процессы репарации ДНК. Из них 83 используются для восстановления 5 типов ДНК, показанных на диаграмме.
Некоторые из наиболее хорошо изученных генов, играющих ключевую роль в этихах восстановления, показаны в таблице. Обозначения генов, показанные красным, серым или голубым цветом, указывают на гены, которые часто эпигенетически изменяются при различных типах рака. Статьи в Википедии о каждом из генов, выделенных красным, серым или голубым цветом, показывают эпигенетические изменения и рак (ы), при обнаруживаются эти эпимутации. В двух обширных экспериментальных обзорных материалах входит большая часть этих эпигенетических недостатков репарации ДНК при раке.
Выделенные или красные гены часто уменьшаются, заглушаются эпигенетическими механизмами при различных формах рака. Когда эти гены имеют экспрессию или ее отсутствие, накапливаться повреждения ДНК. Ошибки репликации после этих повреждений (см. синтез транслезии ) могут привести к увеличению количества мутаций и, в конечном итоге, к раку. Эпигенетическая репрессия генов репарации ДНК в точных путях репарации ДНК, по-видимому, играет центральную роль в канцерогенезе.
Два выделенных серым гена RAD51 и BRCA2 необходимы для гомологичная рекомбинационная репарация. Иногда они эпигенетически чрезмерно выражены, иногда недостаточно выражены при некоторых рака. Как указано стать вях Википедии о RAD51 и BRCA2, такие раковые образования обычно имеют эпигенетические дефекты в других генах репарации ДНК. Эти дефекты репарации, вероятно, вызовут увеличение нереставрированных повреждений ДНК. Сверхэкспрессия RAD51 и BRCA2, наблюдаемая при этих злокачественных новообразованиях, может отражать селективное давление на компенсаторную сверхэкспрессию RAD51 или BRCA2 и повышенную гомологичную рекомбинационную репарацию, чтобы по крайней мере частично справиться с такими избыточными повреждениями ДНК. В тех случаях, когда RAD51 или BRCA2 недоэкспрессированы, это само по себе может приводить к увеличению нерепарированных повреждений ДНК. Ошибки репликации после этих повреждений (см. синтез трансфузии ) могут вызвать усиление мутаций и рак, так что недостаточная экспрессия RAD51 или BRCA2 сама по себе будет канцерогенной.
Выделенные голубым цветом гены находятся в пути соединения концов (MMEJ), опосредованном микрогомологией, активируются при раке. MMEJ - это дополнительный путь неточного ремонта, подверженный ошибкам, для двухцепочечных разрывов. При репарации двухцепочечного разрыва MMEJ гомология 5-25 комплементарных пар оснований между обоими спаренными цепями достаточна для выравнивания цепей, но обычно присутствуют несовпадающие концы (створки). MMEJ удаляет лишние нуклеотиды (створки) в местах соединения цепей, а затем лигирует цепи для создания неповрежденной двойной спирали ДНК. MMEJ почти всегда включает в себя по крайней мере небольшую делецию, так что это мутагенный путь. FEN1, эндонуклеаза лоскута в MMEJ, эпигенетически увеличивается за счет гипометилирования, промотора и сверхэкспрессируется в большинстве случаев рака груди, простата, желудок, нейробластомы, поджелудочная железа и легкие. PARP1 также сверхэкспрессируется, его промоторный участок ETS сайт эпигенетически гипометилирован, и это способствует прогрессированию рака эндометрия, рака яичников с мутацией BRCA и серозного рака яичников с мутацией BRCA. Гены пути MMEJ также сверхэкспрессируются при некоторых раковых заболеваниях (см. MMEJ для обобщения) и также показаны синим цветом.
Дефицит белков репарации ДНК, которые функционируют в точных путях репарации ДНК, увеличивают риск мутации. Скорости мутаций сильно увеличиваются в клетках с мутациями в репарации ошибочного спаривания ДНК или в гомологичной рекомбинационной репарации (HRR). Лица с наследственными мутациями в любом из 34 генов репарации ДНК имеют повышенный риск рака (см. дефекты репарации ДНК и повышенный риск рака ).
При спорадических формах рака дефицита репарации ДНК иногда обнаруживается из-за экспрессии генов репарации ДНК, но гораздо чаще происходит снижение или отсутствие экспрессии генов репарации ДНК происходит из-за эпигенетических изменений, которые уменьшают или молчание экспрессии гена. Например, из 113 случаев прямого рака кишки, исследованных последовательностей, только четыре имели миссенс-мутацию в гене репарации ДНК MGMT, в то время как большинство из них имели пониженную экспрессию MGMT из-за метилирования промотора MGMT. регион (эпигенетическое изменение). Аналогичным образом, из 119 случаев колоректального рака с недостаточной репарацией несоответствие, отсутствующего экспрессия гена репарации ДНК PMS2, белок PMS2 был дефицитным в 6 из-за мутаций в гене PMS2, в то время как в 103 случаях PMS2 была недостаточной, потому что что его партнер по спариванию MLH1 был репрессирован. из-за метилирования промотора (белок PMS2 нестабилен в отсутствии MLH1). В других 10 случаях потеря экспрессии PMS2, вероятно, была связана с эпигенетической сверхэкспрессией микроРНК miR-155, которая подавляет MLH1.
Эпигенетические дефекты в генах репарации ДНК часто встречаются при раке. Таблица множественных видов рака представляет собой частоту, с которой у раковых заболеваний наблюдалась эпигенетическая недостаточность экспрессии гена. Такие эпигенетические дефекты, вероятно, возникают на ранних этапах канцерогенеза, поскольку они также часто появляются (хотя и с несколько меньше) в дефекте поля, окружающем рак, из которого, вероятно, возник рак (см. Таблицу).
| Рак | Ген | Частота при раке | Частота в дефекте поля | Ref. |
|---|---|---|---|---|
| Колоректальный | MGMT | 46% | 34% | |
| Колоректальный | MGMT | 47% | 11% | |
| Колоректальный | MGMT | 70% | 60% | |
| Колоректальный | MSH2 | 13% | 5% | |
| Колоректальный | ERCC1 | 100 % | 40% | |
| Колоректальный | PMS2 | 88% | 50% | |
| Колоректальный | 55% | 40% | ||
| Голова и шея | MGMT | 54% | 38% | |
| Голова и шея | MLH1 | 33% | 25% | |
| Голова и шея | MLH1 | 31% | 20% | |
| Желудок | MGMT | 88% | 78% | |
| Желудок | MLH1 | 73% | 20% | |
| Пищевод | MLH1 | 77 % –100% | 23% –79% |
Похоже, что рак часто может быть инициирован эпигенетическим снижением экспрессии одного или нескольких ферментов репарации ДНК. Снижение репарации ДНК, вероятность накопления повреждений ДНК. Предрасположенный к ошибкам синтез транслезии после некоторых из этих повреждений ДНК может привести к мутации с селективным преимуществом. Клональный патч с избирательным преимуществом может расти и превосходить соседние клетки, образуя полевой дефект. Хотя очевидного селективного преимущества для клетки с пониженной репарацией ДНК не существует, эпимутация гена репарации ДНК может переноситься в качестве пассажира, когда клетки с селективно полезной мутацией тиражируются. В клетках, несущих как эпимутацию гена репарации ДНК, так и мутацию с селективным преимуществом, будут накапливаться дальнейшие повреждения ДНК, которые, в свою очередь, могут привести к дальнейшим мутациям с большими селективными преимуществами. Таким образом, эпигенетические дефекты репарации ДНК в геномах рака и их канцерогенное прогрессирование.
Рак имеет высокий уровень нестабильности генома, связанный с высоким уровнем мутаций. Высокая частота геномных мутаций увеличивает вероятность возникновения мутаций, которые активируют онкогены и инактивируют гены-супрессоры опухоли, что приводит к канцерогенезу. На основе секвенирования всего генома обнаружено, что раковые опухоли содержат от тысяч до сотен тысяч мутаций во всех геномах. (См. Частота мутаций при раке.) Для сравнения также частота мутаций во всем геноме между поколениями людей (от родителей к детям) составляет около 70 новых мутаций на поколение. В областях генома, кодирующих белок, существует всего около 0,35 мутаций между родительскими / дочерними поколениями (менее одного мутировавшего белка на поколение). Секвенирование всего генома в клетках крови пары идентичных близнецов 100-летних долгожителей выявило только 8 соматических различий, хотя соматические вариации, происходящие менее чем в 20% клеток крови, не будут обнаружены.
повреждения ДНК могут вызывать эпигенетические изменения во время неправильных процессов восстановления ДНК. Повреждения ДНК, накапливаются из-за дефектов эпигенетической репарации ДНК, могут быть повышенных эпигенетических изменений, обнаруживаемых во многих генах при раке. В одном из ранних исследований, посвященных ограниченному набору промоторов транскрипции, Fernandez et al. изучили профили метилирования ДНК 855 первичных опухолей. Сравнивая каждый тип опухоли с нормальной тканью, 729 сайтов CpG-островков (55% из 1322 оцененных сайтов CpG) показывают дифференциальное метилирование ДНК. Из этих сайтов 496 были гиперметилированы (репрессированы) и 233 были гипометилированы (активированы). Таким образом, в опухолях наблюдается высокий уровень изменений метилирования эпигенетических промоторов. Некоторые из этих эпигенетических изменений заболевания прогрессирования рака.
Различные соединения эпигенетическими канцерогенами - они вызывают увеличение случаев опухолей, но не проявляют мутаген активность (также следует исключить токсичные соединения или патогены, вызывающие опухоли с повышенной регенерацией). Примеры включают соединения диэтилстильбестрол, арсенит, гексахлорбензол и никель.
Многие тератогены оказывают специфическое воздействие на плод посредством эпигенетических механизмов. Хотя эпигенетические эффекты могут сохранять действие тератогена, такого как диэтилстильбестрол, на протяжении всей жизни пораженного ребенка, возможность врожденных дефектов, возникающих в результате воздействия отцов или у второго и последующих поколений потомства, обычно отвергалась. теоретические основания и отсутствие доказательств. Тем не менее, был продемонстрирован ряд аномалий, опосредованных мужчинами, и, вероятно, их будет больше. В информации на этикетке FDA для Видазы, препарата 5-азацитидина (неметилируемого аналога цитидина, который вызывает гипометилирование при включении в ДНК), указано, что «мужчинам следует рекомендовать не заводить ребенка» при использовании препарата. со ссылкой на доказательства снижения фертильности мышей-самцов, получавших лечение, увеличения потери эмбрионов и аномального развития эмбрионов. У крыс эндокринные различия наблюдались у потомков самцов, подвергшихся воздействию морфина. У мышей были описаны эффекты второго поколения диэтилстильбестерола, возникающие за счет эпигенетических механизмов.
Меланома - это смертельный рак кожи, который возникает из меланоцитов. Известно, что несколько эпигенетических изменений играют роль в переходе меланоцитов в клетки меланомы. Эти изменения являются следствием нарушения регуляции соответствующих ферментов. В число этих ферментов входят несколько гистоновых метилтрансфераз и деметилаз.
Рак простаты ежегодно убивает около 35 000 мужчин, и только в Северной Америке ежегодно диагностируется рак простаты около 220 000 мужчин. Рак простаты является второй по значимости причиной смертельных исходов у мужчин, вызванных раком, и в течение жизни мужчины каждый шестой мужчина болеет этим заболеванием. Нарушения ацетилирования гистонов и метилирования ДНК происходят в различных генах, влияющих на рак простаты. Более 90% при раке предстательной железы подавление гена за счет гиперметилирования CpG-островков промотора GSTP1 гена , который защищает клетки простаты от повреждения генома, вызванного различными окислителями или канцерогенами. Специфическая для метилирования полимеразная цепная реакция (ПЦР) в настоящем времени предполагает, что многие другие гены также гиперметилированы. Экспрессия генов в простате может зависеть от питания и изменения образа жизни.
Второй по распространенности злокачественной опухолью у женщин инвазивный рак шейки матки (ICC) и более 50% всех инвазивного рака шейки матки (ICC) вызывается конгенным вирусом папилломы человека 16 (HPV16 ). Кроме того, интраэпителиальная неоплазия шейки матки (КИН) в первую очередь вызывается онкогенным HPV16. Как и во многих случаях, причинный фактор рака не всегда ведет путь от инфекции к развитию рака. Паттерны геномного метилирования были связаны с инвазивным раком шейки матки. В области HPV16L1 14 протестированных сайтов CpG имеют значительно более высокое метилирование, чем в геномах HPV16 женщин без CIN3. Было обнаружено, что только 2/16 сайтов CpG, протестированных в регуляторной области выше HPV16, связаны с повышенным метилированием в CIN3 +. Это говорит о том, что прямой путь от инфекции к раку иногда сводится к предраковому состоянию при интраэпителиальной неоплазии шейки матки. Кроме того, повышенное метилирование сайта CpG было обнаружено на низких уровнях в большинстве из пяти изученных ядерных генов хозяина, включая 5/5 TERT, 1/4 DAPK1, 2/5 RARB, MAL и CADM1. Кроме того, 1/3 CpG в митохондриальной ДНК были связаны с повышенным метилированием в CIN3 +. Таким образом, существует корреляция между CIN3 + и повышенным метилированием сайтов CpG в открытой рамке считывания L1 HPV16. Это может быть потенциальным биомаркером для будущих скринингов, качественные и предраковые заболевания шейки матки.
Недавние исследования показали, что лейкоз смешанного происхождения <30 Ген>(MLL) вызывает лейкоз путем перестройки и слияния с другими генами в разных хромосомах, процесс находится под эпигенетическим контролем.
Ежегодно в США регистрируется около 15 000 новых случаев саркомы, и около 6200 человек, по прогнозам, умрут от саркомы в США. в 2014 г. Саркомы включают большое количество редких, гистогенетически гетерогенных мезенхимальных опухолей, например, включая хондросаркому, саркому Юинга, лейомиосаркому, липосаркому, остеосаркому, синовиальную саркому и (альвеолярную и эмбриональную) рабдомиосаркому. Некоторые онкогены и гены-супрессоры опухолей в саркомах изменяются эпигенетически. К ним относятся APC, CDKN1A, CDKN2A, CDKN2B, Ezrin, FGFR1, GADD45A, MGMT, STK3, STK4, PTEN, RASSF1A, WIF1, а также несколько микроРНК. Экспрессия эпигенетических модификаторов, таких как компонент BMI1 комплекса PRC1, нарушена при хондросаркоме, саркоме Юинга и остеосаркоме, экспрессия компонента EZH2 комплекса PRC2 нарушена при саркоме Юинга и рабдомиосаркоме. Точно так же экспрессия другого эпигенетического модификатора, гистоновой деметилазы LSD1, увеличивается при хондросаркоме, саркоме Юинга, остеосаркоме и рабдомиосаркоме. Нацеливание на лекарства и ингибирование EZH2 в саркоме Юинга или LSD1 в некоторых саркомах подавляет рост опухолевых клеток в этих саркомах.
Раньше эпигенетические профили были ограничены отдельными генами, находившимися под пристальным вниманием конкретной исследовательской группы. Однако в последнее время ученые переходят к более геномному подходу для определения полного геномного профиля раковых и здоровых клеток.
Популярные подходы к измерению метилирования CpG вках:
Результаты бисульфитного секвенирования обычно подтверждаются с помощью бисульфитного секвенирования [1] с помощью бисульфитного секвенирования CpG, с использованием одного из других методов. Популярные подходы к определению профилей модификаций гистонов в раковых и здоровых клетках включают:
Исследователи надеются определить специфические эпигенетические профили различных типов использования этих профилей в инструментах для более точной диагностики людей. Периодически ученые используют различные варианты эпигеномных профили для определения стадии развития или уровня специфичности у пациентов. Например, гиперметилирование генов, кодирующих (DAPK), p16 и белок эпителиальной мембраны 3 (EMP3), было связано с более агрессивными формами легкого, колоректального и рак мозга. Эти знания могут повлиять на то, как врачи будут ставить диагноз и лечить своих пациентов.
Еще одним фактором, влияющим на лечение пациентов, является знание того, насколько хорошо они будут реагировать на виды лечения. Персонализированные эпигеномные профили раковых клеток пролить свет на эту область. Например, MGMT представляет собой фермент, который обращает добавление алкильных групп к нуклеотиду гуанину. Однако алкилирование гуанина является механизмом, с помощью которого существуют несколько химиотерапевтических препаратов, разрушая ДНК и вызывая гибель клеток. Следовательно, если ген, кодирующий MGMT в раковых клетках, гиперметилирован и фактически подавлен или подавлен, химиотерапевтические препараты, которые путем метилирования гуанина, будут более эффективными, чем в раковых клетках, которые имеют функциональный фермент MGMT.
Эпигенетические биомаркеры также люди в качестве инструментов для молекулярного прогноза. В образцах первичной опухоли и средостения лимфатического узла биопсии гиперметилирование как CDKN2A, так и CDH13 служит маркером для повышенного риска более быстрого рецидива и более высокой смертности пациентов.
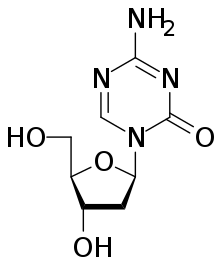 децитабин
децитабин Эпигенетический контроль протоонко-областей и последовательностей супрессоров путем конформационных изменений гистонов играет роль в формировании и прогрессировании рака. Фармацевтические препараты, которые обращают вспять эпигенетические изменения, могут играть роль в различных формах рака.
В последнее время очевидно, что стало известно, что связь между конкретными гистотипами и рака эпигенетическими изменениями может быть разработкой новых эпи-препаратов. Разработка лекарств была сосредоточена в основном на модификации ДНК-метилтрансферазы, гистонацетилтрансферазы (HAT) и гистондеацетилазы (HDAC).
Лекарства, которые специфически нацелены на инвертированный паттерн метилирования раковых клеток, включают ингибиторы ДНК-метилтрансферазы азацитидин и децитабин. Эти гипометилирующие агенты используются для лечения миелодиспластического синдрома, рака крови, вызванного аномальными стволовыми клетками костного мозга. Эти агенты ингибируют все три типа активных ДНК-метилтрансфераз и считались высокотоксичными, но эффективными при использовании в низких дозах, снижая прогрессирование миелодиспластического синдрома до лейкемии.
гистондеацетилазы (Ингибиторы HDAC) проявляют эффективность при использовании Т-клеточной лимфомы. два ингибитора HDAC, вориностат и ромидепсин, были одобрены Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов. Поскольку эти ингибиторы HDAC изменяют состояние ацетилирования многих белков в дополнение к интересующему гистону, требуется знание основного механизма на молекулярном уровне реакции пациента для повышения эффективности использования таких ингибиторов, как лечение. Было обнаружено, что лечение ингибиторами HDAC усиление транскрипции ингибиторами ДНК-метилтрансферазы. Панобиностат одобрен для действий при миеломе.
. Другими фармацевтическими объектами исследования являются гистоновые лизинметилтрансферазы (KMT) и протеин-аргининметилтрансферазы (PRMT). Доклиническое исследование показало, что луназин может иметь благоприятные эпигенетические эффекты.