
Войти
| Восстановление березы | |
|---|---|
| Названо в честь | Артура Берча |
| Тип реакции | Органическая окислительно-восстановительная реакция |
| Идентификаторы | |
| Портал органической химии | восстановление березы |
| RSC ID онтологии | RXNO: 0000042 |
Восстановление березы - это органическая реакция, которая используется для преобразования арены от до циклогексадиенов. Реакция названа в честь австралийского химика Артура Берча. В этом органическом восстановлении ароматических колец в жидком аммиаке с помощью натрия, лития или калия. и спирт, такой как этанол и трет-бутанол. Эта реакция отличается от каталитического гидрирования, которое обычно полностью восстанавливает ароматическое кольцо до циклогексана.

. Примером является восстановление нафталина :

Раствор натрия в жидком аммиаке состоит из соли электрида [Na (NH 3)x] e, которая имеет ярко-синий цвет. сольватированные электроны присоединяются к ароматическому кольцу с образованием анион-радикала. Добавленный спирт поставляет протон радикалу a нион, а также предпоследний карбанион; для большинства субстратов аммиак недостаточно кислый.

Восстановление анизола является одним из простейших примеров и показано в уравнении 1. Восстановление бензойной кислоты показано в уравнении 2.
Место на кольце, где первоначально протонируется анион-радикал, определяет структуру продукта. При использовании донора электронов, такого как метокси (MeO), протонирование алкила некоторыми исследователями рассматривается как орто (т.е. рядом или 1,2) по отношению к заместителю. Другие исследователи полагали, что протонирование происходит в мета (1,3) заместителя. Артур Берч предпочитал мета-протонирование. Считается, что с электроноакцепторными заместителями протонирование происходит в месте заместителя (ipso) или пара (1,4), но это также неясно. Эмпирические правила А. Дж. Берча гласят, что для донорных заместителей конечный продукт имеет максимальное количество заместителей на конечных двойных связях. Для электроноакцепторных групп двойные связи продукта избегают заместителей. Предпочтение размещения групп во время реакции и в конечном продукте называется региоселективностью.

.

Раствор металла в аммиаке дает электроны, которые поглощаются ароматическим кольцом с образованием соответствующего анион-радикала B на первой стадии реакция. За этим следует протонирование спиртом с образованием циклогексадиенильного радикала C. Затем второй электрон переносится на радикал с образованием циклогексадиенилкарбаниона D. На последней стадии второй протон приводит циклогексадиенильный карбанион к неконъюгированному циклогексадиенильный продукт. Эти шаги описаны ниже для анизола.

Известно, что реакция протекает третьего порядка - первого порядка по ароматике, первого порядка по щелочному металлу и первого порядка по спирту. Для этого требуется, чтобы стадия ограничения скорости представляла собой превращение анион-радикала B в циклогексадиенильный радикал C.
Восстановление по Берчу имеет несколько сложных механистических особенностей. Эти особенности определяют региоселективность реакции и рассматриваются ниже. Правило Берча для ароматических соединений с донорами электронов, такими как метоксил или алкил, заключается в том, что продукт будет иметь остаточные двойные связи, несущие максимальное количество заместителей. Для ароматических углеводородов с электроноакцепторными группами, такими как карбоксил, группы заместителей избегают двойных связей. В обоих случаях с электронодонорными и отводящими группами остаточные двойные связи неконъюгированы (см. Ниже). Механизмы реакции, объясняющие эту региоселективность, представляют большой научный интерес. Существенными особенностями являются:
Механизм редукции по Берчу был предметом многочисленных дискуссий. Первоначальный механизм восстановления по Березе включал протонирование анион-радикала, который был мета-метоксигруппой и алкильной группой кольца. Кроме того, было предложено, чтобы последняя стадия, протонирование аниона циклогексадиенила, происходила орто по отношению к этим заместителям. Первоначальный механизм Берча был основан на качественных рассуждениях, а именно, что электронная плотность анион-радикала, возникающая в результате присоединения электрона, будет наивысшей мета-мета для донора электронов (такого как метокси или метил) из-за избегания обычная орто-пара высокая плотность у нейтральных видов.
В 1961 году простые вычисления Хюккеля показали, что предложенный Берчем механизм неверен. Правильный механизм O изображен ниже. Два априорных альтернативных механизма O и M:
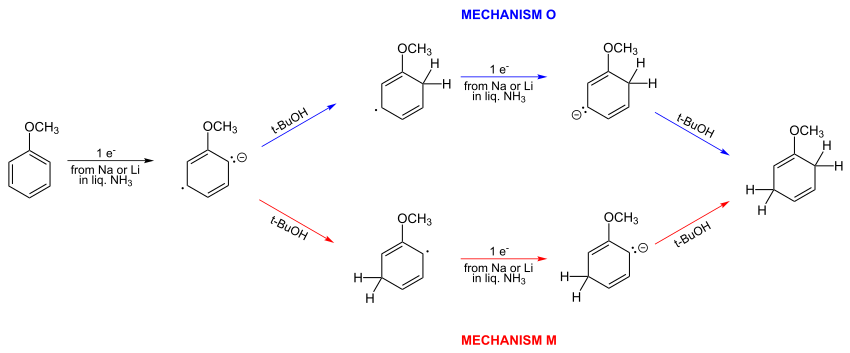
Берч не согласился с этим выводом и продолжил предполагать мета-протонирование анион-радикала. Он предположил, что мета-атака является результатом «противостояния орто- и пара-начального заряда». Ботнер-Би в 1959 году привел качественные аргументы в пользу метапротонирования, как ранее предлагал Берч.
Бернхэм в 1969 году пришел к выводу, что протонирование вряд ли будет происходить преимущественно в орто-положении, и реакция, скорее всего, происходит в мета-положении, но может происходить в обоих местах с одинаковой скоростью.
Впоследствии Берч в обзорной статье отмечалось, что в то время не существовало экспериментального метода, который позволил бы определить правильный механизм. Но он отметил, что публикация Бернхэма благоприятствовала мета-атакам.
В публикациях 1980 года Берч сотрудничал с Лео Радом в исследовании, которое пришло к выводу, что плотности электронов в орто- и мета-положениях близки с небольшим предпочтением орто, но со смесью орто- и мета-положений. происходит протонирование. Ограниченный метод Хартри-Фока на орбитали слейтерского типа (3-g) и неограниченный метод Хартри-Фока слейтер-типа на одном и том же базисном наборе вычислений были использованы для заключения, что и орбитальные, и мета-замены будут происходить с небольшим предпочтением орто.
Затем, в 1990 и 1993 годах, был наконец разработан метод экспериментальной оценки того, протонируют ли анизол и анион-радикал толуола орто или мета. Эзотерический метод начался с предположения, что изотопная селективность при протонировании в среде протий-дейтерий будет больше для анион-радикала первой стадии протонирования, чем для карбаниона предпоследней стадии. Причина заключалась в том, что карбанионы являются гораздо более основными, чем соответствующие анион-радикалы, и поэтому будут реагировать более экзотермически и менее избирательно при протонировании. Экспериментально было определено, что для различных метоксилированных ароматических соединений получается меньше дейтерия в ортоцентре, чем в мета (1: 7). Это следствие большей селективности протонирования анион-радикала. Вычисления (например, ROHF / 6-31g) электронных плотностей совпали с экспериментальными наблюдениями. Также было установлено, что приграничные орбитальные плотности не совпадают, и они использовались в некоторых предыдущих отчетах.
Впоследствии, в 1992 и 1996 годах, Берч дважды публиковал материалы, по-прежнему предлагая предпочтение метапротонированию. Это было изменением его более ранних взглядов, опубликованных вместе с Лео Радомом.
Однако в учебниках, посвященных механизму восстановления по Березе, отмечается, что орто-протонирование исходного анион-радикала является предпочтительным.
В отличие от примеров с электронодонорными заместителями, случай с акцептирующими группами более очевиден. Таким образом, как показано ниже, структура предпоследнего дианиона D характеризуется тем, что он подвержен улавливанию алкилгалогенидами.
Механизм восстановления бензойных кислот, включая возможное алкилирование.

Этот дианион возникает независимо от того, используется ли спирт при восстановлении или нет. Таким образом, начальное протонирование трет-бутиловым спиртом или аммиаком является парами, а не ipso, как видно на стадии от B до C.
Вторая стадия восстановления по Берчу с получением неконъюгированных циклогексадиенов также ставит механистические вопросы. Таким образом, как показано на рисунке ниже, для карбаниона существуют три резонансные структуры B, C и D. Простые вычисления Хюккеля приводят, как отмечено в первой записи таблицы ниже, к равным электронным плотностям у трех атомов 1, 3 и 5. Однако, в отличие от плотностей, вычисление Хюккеля менее наивно в отношении порядков связей, а связи 2–3 и 5–6 будут сокращены, как показано в первой записи таблицы. С помощью порядков связи, изменяющих простые обменные интегралы, в вычислении Малликена-Веланда-Манна было показано, что плотность электронов в центральном атоме 1 становится наибольшей. Более современные вычисления RHF приводят к тому же результату.
Введение электрона в бензол и 3 резонансные структуры для карбаниона второй стадии и центральное протонирование с образованием несопряженного диена:

Пять атомов углерода циклогексадиенильного аниона.
| Приближение | Атом плотности 3 | Атом плотности 2 | Атом плотности 1 | Порядок связывания 2–3 | Связь Порядок 1-2 |
|---|---|---|---|---|---|
| Hückel (1-е прибл.) | 0,333 | 0,00 | 0,333 | 0,788 | 0,578 |
| 2-е приблизительно | 0,317 | 0,00 | 0,365 | 0,802 | 0,564 |
| 3-е прибл. | 0,316 | 0,00 | 0,368 | 0,802 | 0,562 |
Существуют известные прецеденты протонирования центрального аниона. Таким образом, конъюгированные еноляты как C = CC = CO- в течение некоторого времени были известны как кинетически протонирующие в центре енолятной системы с получением β, γ-ненасыщенного карбонильного соединения в условиях, когда анион, а не енол, является разновидностью протонированный.
В присутствии алкилгалогенида карбанион также может подвергаться нуклеофильному замещению на образование углерод-углеродной связи. В замещенных ароматических соединениях электроноакцепторный заместитель, такой как карбоновая кислота, стабилизирует карбанион, а наименее замещенный олефин представляет собой генерируется. С электронодонорным заместителем достигается противоположный эффект. В результате реакции образуется больше менее термодинамически стабильного несопряженного 1,4-аддитивного продукта, чем более стабильного сопряженного 1,3-диена, поскольку наибольший орбитальный коэффициент ВЗМО промежуточного соединения сопряженного пентадиенильного аниона центральный атом углерода. После образования образующийся 1,4-циклогексадиен не может уравновеситься с термодинамически более стабильным продуктом; следовательно, образуется наблюдаемый кинетический продукт. Существуют также экспериментальные альтернативы щелочным металлам, которые более безопасны в обращении, такие как восстановитель M-SG.
При алкилировании березы анион, образованный при восстановлении по Берчу, улавливается подходящим электрофилом, таким как галогеналкан, например:

. В реакции, изображенной ниже, 1,4-дибромбутан добавляют к трет-бутилбензоату с образованием алкилированного 1,4-циклогексадиенового продукта:

Поскольку жидкий аммиак имеет чтобы конденсироваться в колбе и испариться в течение ночи после завершения реакции, вся процедура может быть довольно хлопотной и трудоемкой. Однако использовались альтернативные растворители, такие как THF, а также смесь н-пропиламина и этилендиамина, оба с сопоставимыми результатами. Последний на самом деле является модификацией реакции Бенкезера, которая в ее первоначальных формах имеет тенденцию полностью восстанавливать нафталин до октагидро- и декагидронафталина.
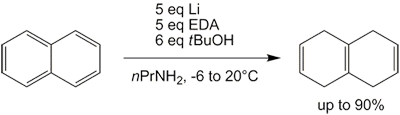
В результате восстановления нафталина до изотетралина (1,4,5,8-тетрагидронафталина) в качестве побочного продукта образуется тетралин (1,2,3,4-тетрагидронафталин), как и в случае с обычным Сокращение березы.
Восстановление может осуществляться за счет внешнего потенциала жертвенного анода (магния или алюминия). Диметилмочевина, донор протонов, стабилизирует промежуточное соединение ионами лития (из солей).
Эта реакция приписывается Артуру Берчу (1915–1995), когда он работал в лаборатории Дайсона Перринса в университете . of Oxford, основанный на более ранней работе Вустера и Годфри, опубликованной в 1937 году. Он превращает ароматические соединения, имеющие бензоидное кольцо, в продукт, 1,4-циклогексадиены, в котором два атома водорода присоединены к противоположным концам молекулы.
В исходной реакции, описанной Берчем в 1944 г., использовались натрий и этанол. Альфред Л. Уайлдс позже обнаружил, что литий дает более высокие выходы.