
Войти
A биоподобный - это биологический медицинский продукт (также известный как биологический) очень похож на другой уже одобренный биологический препарат («эталонный препарат»). Биосимиляры одобрены в соответствии с теми же стандартами фармацевтического качества, безопасности и эффективности, которые применяются ко всем биологическим лекарствам. Биосимиляры являются официально одобренными версиями оригинальных «новаторских» продуктов и могут быть изготовлены по истечении срока действия патента на исходный продукт. Ссылка на новаторский продукт является неотъемлемой частью одобрения.
В отличие от генериков более распространенного низкомолекулярного типа, биопрепараты обычно обладают высокой молекулярной сложностью и могут быть весьма чувствительны к изменениям в производственных процессах. Несмотря на эту неоднородность, все биофармацевтические препараты, включая биоаналоги, должны поддерживать постоянное качество и клиническую эффективность на протяжении всего своего жизненного цикла. Биоаналог не рассматривается как генерик биологического лекарства. В основном это связано с тем, что естественная изменчивость и более сложное производство биологических лекарств не позволяют точно воспроизвести молекулярную микрогетерогенность. Органы, связанные с лекарствами, такие как Европейское агентство по лекарственным средствам (EMA) ЕС, Управление по контролю за продуктами и лекарствами (FDA) и Отделение продуктов здравоохранения и продуктов питания из Министерства здравоохранения Канады имеют собственное руководство по требованиям к демонстрации сходства двух биологических продуктов с точки зрения безопасности и эффективности. По их мнению, аналитические исследования демонстрируют, что биологический продукт очень похож на эталонный продукт, несмотря на незначительные различия в клинически неактивных компонентах, исследованиях на животных (включая оценку токсичности) и клинических исследованиях (включая оценку <135.>иммуногенность и фармакокинетика или фармакодинамика ). Их достаточно, чтобы продемонстрировать безопасность, чистоту и эффективность в одном или нескольких подходящих условиях использования, для которых эталонный продукт лицензирован и предназначен для использования, и для которых запрашивается лицензия на биологический продукт.
Всемирная организация здравоохранения (ВОЗ) опубликовала «Руководство по оценке аналогичных биотерапевтических продуктов (SBP)» в 2009 году. Цель этого руководства - предоставить международную норму для оценки биоаналоги с высокой степенью сходства с уже лицензированным эталонным биотерапевтическим препаратом.
Европейский Союз был первым регионом в мире, который разработал правовую, нормативную и научную базу для утверждения биоаналогичных препаратов. С 2006 года EMA выдало разрешение на продажу более 50 биоподобных препаратов (первый одобренный биоаналог соматропина (гормона роста)). Первым биоаналогом моноклонального антитела, который был одобрен во всем мире, был биоаналог инфликсимаба в ЕС в 2013 году. 6 марта 2015 года FDA одобрило первый биоподобный продукт в США, биоподобный препарат филграстим, названный филграстим-sndz (торговое название Zarxio) от Sandoz.
Утверждение лекарственных средств в ЕС опирается на прочную правовую базу, которая в 2004 году ввела специальный маршрут для утверждения биосимиляров. ЕС стал пионером в регулировании биосимиляров с момента утверждения первого из них (гормона роста соматропина) в 2006 году. С тех пор ЕС одобрил наибольшее количество биоподобных препаратов в мире и, следовательно, имеет самый обширный опыт их использования и безопасности.. Все лекарства, производимые с использованием биотехнологий и для конкретных показаний (например, для лечения рака, нейродегенерации и аутоиммунных заболеваний), должны быть одобрены в ЕС через EMA (через так называемую «централизованную процедуру»). Почти все биосимиляры, одобренные для использования в ЕС, были одобрены централизованно, поскольку для их производства используются биотехнологии. Некоторые биоаналоги могут быть одобрены на национальном уровне, например, некоторые низкомолекулярные гепарины, полученные из слизистой оболочки кишечника свиней. Когда компания подает заявку на получение регистрационного удостоверения в EMA, данные оцениваются научными комитетами EMA по лекарственным средствам для человека и безопасности (CHMP и PRAC), а также экспертами ЕС по биологическим лекарствам (Рабочая группа по биологическим препаратам) и специалистами по биосимилярам (Biosimilar). Рабочая группа). По результатам проверки EMA вырабатывается научное заключение, которое затем отправляется в Европейскую комиссию, которая в конечном итоге дает разрешение на маркетинг в масштабах всего ЕС.
В США Управление по контролю за продуктами и лекарствами (FDA) постановило, что необходимо новое законодательство, чтобы позволить им утверждать биосимиляры тем биологическим препаратам, которые были первоначально одобрены в рамках закона PHS. Дополнительные слушания в Конгрессе были проведены. 17 марта 2009 г. в Палате представителей был представлен Закон о путях получения биосимиляров. См. веб-сайт Библиотеки Конгресса и выполните поиск по HR 1548 на 111-й сессии Конгресса. С 2004 года FDA провело серию общественных встреч по биосимилярам.
FDA получило полномочия одобрять биоподобные препараты (включая взаимозаменяемые, заменяемые их референсным продуктом) в рамках Защиты пациентов и доступных Закон об уходе подписан президентом Обамой 23 марта 2010 года.
FDA ранее одобряло биологические продукты с использованием сопоставимости, например, Omnitrope в мае 2006 года, но это похоже на Эноксапарин также относился к препарату сравнения, Генотропин, первоначально одобренному в качестве препарата согласно Закону FDC.
6 марта 2015 г. Zarxio получил первое одобрение FDA. Zarxio от Sandoz является биологическим аналогом Neupogen (филграстим) компании Amgen, который был первоначально лицензирован в 1991 году. Это первый продукт, который был принят в соответствии с Законом о ценовой конкуренции и инновациях в биопрепаратах 2009 года (BPCI Act), который был принят в рамках Закона о доступном здравоохранении. Закон. Но Zarxio был одобрен как биоаналог, а не как взаимозаменяемый продукт, отмечает FDA. И согласно Закону BPCI только биологический препарат, который был одобрен как «взаимозаменяемый», может быть заменен эталонным продуктом без вмешательства поставщика медицинских услуг, прописавшего эталонный продукт. FDA заявило, что одобрение Zarxio основано на обзоре доказательств, включающих структурные и функциональные характеристики, данные исследований на животных, данные фармакокинетики и фармакодинамики человека, данные клинической иммуногенности и другие данные о клинической безопасности и эффективности, которые демонстрируют, что Zarxio является биоподобен нейпогену.
В марте 2020 года большинство белковых продуктов, которые были одобрены в качестве лекарственных препаратов (включая все инсулины, представленные в настоящее время на рынке по состоянию на декабрь 2019 года), планируется открыть для конкуренции с биоподобными и взаимозаменяемыми продуктами в США. Однако «химически синтезированные полипептиды» исключены из этого перехода, что означает, что продукт, попадающий в эту категорию, не сможет поступить на рынок как биоподобный или взаимозаменяемый продукт, а должен будет поступать на рынок под другим
Клонирование генетического материала человека и разработка систем биологического продуцирования in vitro позволили получить практически любое биологическое вещество на основе рекомбинантной ДНК для возможной разработки препарат. Технология моноклональных антител в сочетании с технологией рекомбинантной ДНК проложила путь для создания индивидуальных и целевых лекарств. Гены- и клеточная терапия появляются как новые подходы.
Рекомбинантные терапевтические белки имеют сложную природу (состоят из длинной цепочки аминокислот, модифицированных аминокислот, дериватизированных фрагментами сахара, сложенных сложными механизмами). Эти белки производятся в живых клетках (линиях клеток бактерий, дрожжей, животных или человека). Конечные характеристики лекарственного средства, содержащего рекомбинантный терапевтический белок, в значительной степени определяются процессом, посредством которого они производятся: выбором типа клетки, разработкой генетически модифицированной клетки для производства, производственным процессом, процессом очистки, формулированием терапевтический белок в лекарство.
После истечения срока действия патента на одобренные рекомбинантные препараты (например, инсулин, человеческий гормон роста, интерфероны, эритропоэтин, моноклональные антитела и т. Д.) Любая другая биотехнологическая компания может разработать и продать эти биопрепараты (так называемые биоподобные препараты). Каждый биологический (или биофармацевтический продукт) демонстрирует определенную степень вариабельности даже между разными партиями одного и того же продукта, что связано с присущей вариативностью системы биологической экспрессии и производственного процесса. Любой вид эталонного продукта претерпел многочисленные изменения в производственных процессах, и такие изменения в производственном процессе (от смены поставщика сред для культивирования клеток до новых методов очистки или новых производственных площадок) были подтверждены соответствующими данными и одобрены. EMA. Напротив, биосимиляры обязательно должны пройти как доклинические, так и клинические испытания, которые должны продемонстрировать наиболее чувствительные клинические модели, чтобы выявить различия между двумя продуктами с точки зрения человеческой фармакокинетики (PK) и фармакодинамика (PD), эффективность, безопасность и иммуногенность.
Современная концепция разработки биоподобных моноклональных антител следует принципу, согласно которому обширное физико-химическое, аналитическое и функциональное сравнение молекул дополняется сравнительными доклиническими и клиническими данными, которые устанавливают эквивалентную эффективность и безопасность в клиническое «модельное» показание, которое наиболее чувствительно для обнаружения любых незначительных различий (если они существуют) между биоаналогом и его эталонным mAb, также на клиническом уровне.
Европейское агентство по лекарственным средствам (EMA) признало этот факт, что привело к введению термина «биоподобный» в знак признания того факта, что, хотя биоподобные продукты похожи на исходный продукт, они не совсем то же самое. Каждый биологический объект демонстрирует определенную степень изменчивости. Однако при условии, что структура и функции, фармакокинетические профили и фармакодинамический эффект (ы) и / или эффективность могут быть сопоставимы для биоподобного и эталонного продукта, те побочные реакции на лекарства, которые связаны с преувеличенными фармакологическими эффектами, также могут можно ожидать на аналогичных частотах.
Первоначально сложность биологических молекул привела к запросам о существенных данных об эффективности и безопасности для утверждения биоподобных препаратов. Это постепенно заменяется большей зависимостью от анализов, от качественных до клинических, которые демонстрируют чувствительность анализа, достаточную для обнаружения любых значительных различий в дозах. Однако безопасное применение биопрепаратов зависит от информированного и надлежащего использования медицинскими работниками и пациентами. Внедрение биосимиляров также требует специально разработанного плана фармаконадзора. Воссоздать биопрепараты сложно и дорого, потому что сложные белки получены из генетически модифицированных живых организмов. Напротив, низкомолекулярные препараты, состоящие из соединений на химической основе, могут быть легко воспроизведены, и их воспроизведение значительно дешевле. Для того, чтобы быть опубликованным, биоподобия должны быть продемонстрированы как близкие к идентичным исходному биологическому продукту-изобретателю на основании данных, собранных в ходе клинических, животных, аналитических исследований и конформационного статуса.
Как правило, один раз Препарат выпускается на рынок FDA, его безопасность и эффективность необходимо повторно оценивать каждые шесть месяцев в течение первого и второго года. После этого ежегодно проводятся повторные оценки, и о результатах оценки следует сообщать в такие органы, как FDA. Биосимиляры должны пройти нормативы фармаконадзора (PVG) в качестве эталонного продукта. Таким образом, биосимиляры, одобренные EMA (Европейским агентством по лекарственным средствам), должны представлять план управления рисками (RMP) вместе с маркетинговым заявлением и должны предоставлять регулярные отчеты о безопасности после того, как продукт появится на рынке. RMP включает профиль безопасности препарата и предлагает проспективные исследования фармаконадзора.
Несколько исследований PK, таких как исследования, проведенные Комитетом по лекарственным средствам для человека (CHMP), были проведены в различных диапазонах условий; Антитела от оригинального продукта против антител от биоаналога; комбинированная терапия и монотерапия; различные заболевания и т. д. с целью проверки сравнимости фармакокинетики биосимиляра с эталонным лекарственным средством в достаточно чувствительной и однородной популяции. Важно отметить, что при условии, что структура и функции, фармакокинетические профили и фармакодинамический эффект (-ы) и / или эффективность могут быть сопоставимы для биоаналога и эталонного продукта, те побочные реакции на лекарства, которые связаны с преувеличенными фармакологическими эффектами, также могут можно ожидать на аналогичных частотах.
На сегодняшний день в Европейском Союзе находится наибольшее количество одобренных биосимиляров. Научные комитеты EMA оценивают большинство заявок на регистрацию биосимиляров перед тем, как они могут быть одобрены и проданы в ЕС. EMA оценивает биосимиляры в соответствии с теми же стандартами фармацевтического качества, безопасности и эффективности, которые применяются ко всем биологическим лекарствам, одобренным в ЕС.
| Действующее вещество | Эталонный препарат | Биосимиляры |
|---|---|---|
| Адалимумаб (8) | Хумира | Амгевита, Амспарити, Кадалимаб Халиматоз, Хефия, Хулио, Хиримос, Идацио, Имральди |
| Бевацизумаб (4) | Авастин | Мваси, Зирабев, Айбинтио Эквидасент |
| Эноксапарин натрия (1) | Ловенокс | Инхикса |
| Эпоэтин альфа (5) | Эпрекс / Erypo | Abseamed, Binocrit, Epoetin Alfa Hexal, Retacrit, Silapo |
| Etanercept (3) | Enbrel | Benepali, Erelzi, Nepexto |
| Filgrastim (7) | Нейпоген | Аккофил, Филграстим Гексал, Грастофил, Нивестим, Ратиограстим, Теваграстим, Зарцио |
| Фоллитропин альфа (2) | Гонал-Ф | Бемфола, Овалеп |
| Инфликсимаб (4) | 262>Flixabi, Inflectra, Remsima, Zessly | |
| Инсулин аспарт (1) | НовоРапид | Инсулин аспарт Санофи |
| Инсулин гларгин (2) | Лантус | Абасаглар, Семгли |
| Инсулин лиспро (1) | Хумалог | Инсулин лиспро Санофи |
| Пегфилграстим (7) | Неуласта | Цегфила, Фулфила, Грасустек, Пелграз, Пельмег, Уденика, Зикстензо |
| Ритуксимаб (6) | Мабтера | Блицима, Ритемвиа, Риксатон, Риксимио, Руксиенс, Труксима |
| Соматропин (1) | Генотропин | Омнитроп |
| Терипара | Forsteo | Movymia, Terrosa |
| Trastuzumab (6) | Herceptin | Herzuma, Kanjinti, Ogivri, Ontruzant, Trazimera, Zercepac |
Источник: Европейское агентство по лекарственным средствам (июнь 2020 г.) https://www.biosimilars-nederland.nl/wp-content/uploads/2020_06_01-Table-EU-licensed-biosimilars-by-molecule_May_2020agv.pdf
Закон о ценовой конкуренции и инновациях в биопрепаратах от 2009 года (Закон BPCI) был первоначально спонсирован и внесен 26 июня 2007 года сенатором Эдвардом Кеннеди (штат Массачусетс). Он был официально принят в соответствии с Законом о защите пациентов и доступном медицинском обслуживании (Закон PPAC), подписанным президентом Бараком Обамой 23 марта 2010 года. Закон BPCI был поправкой к Закону о государственной службе здравоохранения (PHS). Act) для создания сокращенного пути утверждения биологических продуктов, которые продемонстрировали высокую степень сходства (биоподобия) с биологическим продуктом, одобренным Управлением по контролю за продуктами и лекарствами (FDA). Закон BPCI концептуально аналогичен Закону о конкуренции цен на лекарства и восстановлении срока действия патентов 1984 года (также именуемому «Закон Хэтча-Ваксмана»), который разрешил биологические препараты через Федеральный закон о пищевых продуктах, лекарствах и косметических средствах (FFDC. Закон). Закон BPCI согласуется с давней политикой FDA, разрешающей надлежащее использование того, что уже известно о лекарстве, тем самым экономя время и ресурсы и избегая ненужного дублирования испытаний на людях или животных. FDA выпустило в общей сложности четыре проекта руководящих принципов, касающихся биоподобных или последующих биологических препаратов. После выпуска первых трех руководящих документов FDA провело публичные слушания 11 мая 2012 года.
В 2018 году FDA выпустило План действий по биосимилярам для выполнения положений BPCI, включая ограничение злоупотребления Система оценки и снижения рисков (REMS) для вечнозеленого и перевода инсулина и гормона роста человека на регуляцию в качестве биологических препаратов, а не лекарств.
| Дата утверждения биосимиляра FDA | Биоподобный продукт | Оригинальный продукт |
|---|---|---|
| 6 марта 2015 г. | filgrastim-sndz / Zarxio | filgrastim / Neupogen |
| 5 апреля 2016 г. | инфликсимаб-дийб / Инфлектра | инфликсимаб / Ремикейд |
| 30 августа 2016 г. | этанерцепт-szzs / Эрелци | этанерцепт / Энбрел |
| сентябрь 23, 2016 | адалимумаб-атто / Амджевита | адалимумаб / Хумира |
| 21 апреля 2017 г. | инфликсимаб-абда / Renflexis | инфликсимаб / Remicade |
| 25 августа 2017 г. | адалимумаб-адбм / цилтезо | адалим umab / Humira |
| 14 сентября 2017 г. | bevacizumab-awwb / Mvasi | bevacizumab / Avastin |
| 1 декабря 2017 г. | trastuzumab-dkst / Огиври | трастузумаб / Герцептин |
| 13 декабря 2017 г. | инфликсимаб-qbtx / Иксифи | инфликсимаб / Ремикейд |
| 15 мая 2018 г. | эпоэтин альфа-epbx / Retacrit | эпоэтин альфа / Procrit |
| 4 июня 2018 г. | pegfilgrastim-jmdb / Fulphila | pegfilgrastim / Neulasta |
| 20 июля 2018 г. | филграстим-аафи / Нивестим | филграстим / Нейпоген |
| 30 октября 2018 г. | адалимумаб-адаз / Хиримоз | адалимумаб / Хумира |
| 2 ноября 2018 г. | пегфилграстим-cbqv / Udenyca | пегфилграстим / Neulasta |
| 28 ноября 2018 г. | ритуксимаб-abbs / Truxima | ритуксимаб / Ритуксан |
| 14 декабря 2018 г. | трастузумаб-пкрб / Герзума | трастузумаб / Герцептин |
| 18 января 2019 г. | трастузумаб-дттб / Онтрузант | трастузумаб / Герцептин |
| 11 марта 2019 г. | трастузумаб-гуйп / Тразимера | трастузумаб / Герцептин |
| 25 апреля 2019 г. | этанерцепт-икро / Этиково | этанерцепт / Энбрел |
| 13 июня 2019 г. | трастузумаб- anns / Канджинти | трастузумаб / Герцептин |
| 27 июня 2019 г. | бевацизумаб-бвзр / Зирабев | бевацизумаб / Авастин |
| 23 июля 2019 г. | ритуксимаб-пввр / Ruxience | ритуксимаб / Rituxan |
| 23 июля 2019 г. | адалимумаб-bwwd / Hadlima | адалимумаб / Humira |
| 4 ноября 2019 г. | pegfilgrastim-bmez / Ziextenzo | pegfilgrastim / Neulasta |
| 15 ноября 2019 г. | адалимумаб-afzb / Abrilada | адалимумаб / Humira |
| 6 декабря 2019 г. | инфликсимаб-axxq / Avsola | инфликсимаб / Remicade |
| 10 июня 2020 г. | pegfilgrastim-apgf / Nyvepria | pegfilgrastim / Neulasta |
В Европе не требуется уникальный идентификатор биоподобного лекарственного препарата, соблюдаются те же правила, что и для всех биопрепаратов. Для идентификации и отслеживания биологических препаратов в ЕС, лекарства должны отличаться торговым наименованием и номером партии, и это особенно важно в случаях, когда на рынке присутствует более одного лекарства с одинаковым МНН. Это гарантирует, что в соответствии с требованиями ЕС к отчетности о нежелательных реакциях лекарственного средства лекарство может быть правильно идентифицировано в случае возникновения каких-либо проблем с безопасностью (или иммуногенностью) конкретного продукта. В отчете 1 Консультации экспертов ВОЗ по расширению доступа к аналогичным биотерапевтическим продуктам и их использованию, опубликованном в октябре 2017 г., говорится на странице 4, что после итогов встречи: «Не было достигнуто консенсуса относительно того, следует ли ВОЗ продолжать с BQ... ВОЗ в настоящее время этим заниматься не будет ". 14 февраля 2019 года Министерство здравоохранения Канады объявило о решении использовать как торговую марку, так и непатентованное наименование в процессе использования лекарств. Биопрепараты с одним и тем же непатентованным названием можно отличить по уникальным торговым маркам. США выбрали другой подход, поскольку они являются единственной юрисдикцией, требующей присвоения четырехзначного буквенного суффикса непатентованному наименованию оригинального продукта, чтобы различать лекарственные средства-новаторы и их биоаналоги.
В Соединенных Штатах биосимиляры не оказали ожидаемого влияния на цены, что привело к предложению 2019 года о регулировании цен после периода эксклюзивности. Другое предложение требует, чтобы создатели совместно использовали лежащие в основе клеточные линии.
. В 2019 году предложенный Закон о прозрачности биологических патентов поможет решить проблему вечнозеленых «патентных зарослей », требуя, чтобы все патенты раскрытие информации о защите биоподобных веществ.
Биосимиляры столкнулись с трудностями при получении доли на рынке, что вынудило разработчика биосимиляров Pfizer подать в суд на Johnson Johnson из-за антиконкурентных контрактов с менеджеры по льготам аптек, которые объединяют скидки; их иногда называют «стеной скидок», и скидки, как правило, недоступны для клиентов.
Предлагаемое правило, влияющее на участников программ Medicare / Medicaid, объявленное позже в 2019 г. Предлагаемый закон под названием «Закон о ценах по рецепту для людей» от 2019 г. представил запрос, чтобы FTC расследовала скидки. В 2019 году руководители фармацевтических компаний дали показания перед комитетом Сената, а компании не согласились с реформой конкуренции в области биоподобных препаратов. Комитет по надзору Палаты представителей и Финансовый комитет Сената провели слушания в начале 2019 года.
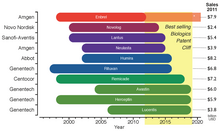 Патентный обрыв 2012–2019 гг.. Период эксклюзивности рынка до даты истечения срока действия патента для десяти самых продаваемых биопрепаратов в 2011 году. * Enbrel получила одобрение в 2011 году на патент, поданный в 1995 году, что продлило срок его действия еще на 17 лет.
Патентный обрыв 2012–2019 гг.. Период эксклюзивности рынка до даты истечения срока действия патента для десяти самых продаваемых биопрепаратов в 2011 году. * Enbrel получила одобрение в 2011 году на патент, поданный в 1995 году, что продлило срок его действия еще на 17 лет. Законодательные требования Пути утверждения, вместе с дорогостоящими производственными процессами, увеличивают затраты на разработку биоподобных препаратов, которые могут составлять от 75 до 250 миллионов долларов за молекулу. Этот барьер для выхода на рынок влияет не только на компании, желающие их производить, но также может задерживать доступность недорогих альтернатив для государственных медицинских учреждений, которые субсидируют лечение своих пациентов. Несмотря на то, что рынок биосимиляров растет, падение цен на биологические препараты с риском истечения срока действия патента не будет таким большим, как на другие генерические препараты; на самом деле было подсчитано, что цена на биоподобные продукты будет составлять 65-85% от их оригинальных. Биосимиляры привлекают внимание рынка, поскольку грядет патентный обрыв, который поставит под угрозу почти 36% рынка биологических препаратов стоимостью 140 миллиардов долларов (по состоянию на 2011 год), учитывая только 10 самых продаваемых продуктов.
Мировой рынок биоподобных препаратов составил 1,3 миллиарда долларов США в 2013 году и, как ожидается, достигнет 35 миллиардов долларов США к 2020 году за счет истечения срока действия патента дополнительных десяти биологических препаратов-блокбастеров.
Некоторые компании (в некоторых случаях дочерние компании), как правило, действуют как производители непатентованных лекарств, при этом крупные из них, включая Teva, Mylan и Sandoz, могут также распространите это внимание на биоаналоги. Сандоз, например, представил первый биоаналог в США и планирует выпустить еще один в 2020 году. Новые компании, такие как индийская Cadila Pharmaceuticals, Sun Pharma, Aurobindo Pharma и Dr. Reddy's Laboratories, а также базирующаяся в Канаде Apotex приобрели долю традиционных дженериков, что побудило старые компании переключить свое внимание на сложные лекарства, такие как биоподобные препараты.