
Войти
| Автор (ы) | Chapman B, Chang J |
|---|---|
| Первоначальный выпуск | 2000 (2000) |
| Стабильный выпуск | 1.74 / 16 июля 2019 г.; 15 месяцев назад (2019-07-16) |
| Репозиторий | https://github.com/biopython/biopython |
| Написано на | Python и C |
| Platform | Cross- платформа |
| Тип | Биоинформатика |
| Лицензия | Лицензия на Biopython |
| Веб-сайт | biopython.org |
Проект Biopython является открытым source коллекция некоммерческих Python инструментов для вычислительной биологии и биоинформатики, созданная международной ассоциацией разработчиков. Он содержит классы для представления биологических последовательностей и аннотаций последовательностей, и он может читать и записывать в файлы различных форматов. Он также позволяет использовать программные средства доступа к онлайновым базам данных биологической информации, например, в NCBI. Отдельные модули расширяют возможности Biopython для выравнивания последовательностей, структуры белка, популяционной генетики, филогенетики, мотивов последовательностей, и машинное обучение. Biopython - один из нескольких проектов Bio *, направленных на сокращение дублирования кода в вычислительной биологии.
Разработка Biopython началась в 1999 году и впервые была выпущена в июле 2000 года. в течение аналогичного периода времени и с аналогичными целями для других проектов, которые добавили возможности биоинформатики в свои соответствующие языки программирования, включая BioPerl, BioRuby и BioJava. Первыми разработчиками проекта были Джефф Чанг, Эндрю Далке и Брэд Чепмен, хотя на сегодняшний день внесли свой вклад более 100 человек. В 2007 году был создан аналогичный проект Python, а именно PyCogent .
Первоначальная сфера применения Biopython заключалась в доступе, индексировании и обработке файлов биологических последовательностей. Хотя это по-прежнему является основным направлением, в последующие годы добавленные модули расширили его функциональность, чтобы охватить дополнительные области биологии (см. Ключевые особенности и примеры).
Начиная с версии 1.62, Biopython поддерживает работу как на Python 3, так и на Python 2.
Везде, где это возможно, Biopython следует соглашениям, используемым языком программирования Python для облегчить пользователям, знакомым с Python. Например, объектами Seqи SeqRecordможно управлять с помощью нарезки аналогично строкам и спискам Python. Он также спроектирован так, чтобы быть функционально похожим на другие проекты Bio *, такие как BioPerl.
Biopython может читать и записывать наиболее распространенные форматы файлов для каждой из своих функциональных областей, а его лицензия разрешает и совместима с большинство других лицензий на программное обеспечение, которые позволяют использовать Biopython в различных программных проектах.
Основной концепцией Biopython является биологическая последовательность, и это представлено классом Seq. Объект Biopython Seqво многих отношениях похож на строку Python: он поддерживает нотацию фрагментов Python, может быть объединен с другими последовательностями и является неизменяемым. Кроме того, он включает методы, специфичные для последовательности, и определяет конкретный используемый биологический алфавит.
>>># Этот сценарий создает последовательность ДНК и выполняет некоторые типичные манипуляции>>>из Bio.Seq import Seq>>>из Bio.Alphabet import IUPAC>>>dna_sequence = Seq ('AGGCTTCTCGTA', IUPAC.unambiguous_dna)>>>dna_sequence Seq ('AGGCTTCTCGTA', IUPACUnambiguousDNA ())>>>dna_sequence [2: 7] Seq ('GCTTC', IUPACUnambiguousDNA ())>>>dna_sequence.reverse_complement () Seq ('TACGAGAAGCCUTACT))>>>rna_sequence = dna_sequence.transcribe ()>>>rna_sequence Seq ('AGGCUUCUCGUA', IUPACUnambiguousRNA ())>>>rna_sequence.translate () Seq ('RLLV', IUPACPotein ())Класс SeqRecordописывает последовательности вместе с такой информацией, как имя, описание и особенности в форме объектов SeqFeature. Каждый объект SeqFeatureопределяет тип функции и ее расположение. Типы признаков могут быть «ген», «CDS» (кодирующая последовательность), «repeat_region», «mobile_element» или другие, а положение признаков в последовательности может быть точным или приблизительным.
>>># Этот скрипт загружает аннотированную последовательность из файла и просматривает часть ее содержимого.>>>из Bio import SeqIO>>>seq_record = SeqIO.read ('pTC2.gb', 'genbank')>>>seq_record.name 'NC_019375'>>>seq_record.description 'Providencia stuartii плазмида pTC2, полная последовательность. '>>>seq_record.features [14] SeqFeature (FeatureLocation (ExactPosition (4516), ExactPosition (5336), strand = 1), type = 'mobile_element')>>>seq_record.seq Seq ('GGATTGAATATAACCGACGTGACTGTCTACATTTAGCTAGACGACTGACTGTCTACATGCCCGACGAC IUPACAmbiguousDNA ())Biopython может читать и записывать в ряд распространенных форматов последовательностей, включая FASTA, FASTQ, GenBank, Clustal, ФИЛИП и НЕКСУС. При чтении файлов описательная информация в файле используется для заполнения членов классов Biopython, таких как SeqRecord. Это позволяет преобразовывать записи одного формата файла в другие.
Очень большие файлы последовательностей могут превышать ресурсы памяти компьютера, поэтому Biopython предоставляет различные варианты доступа к записям в больших файлах. Они могут быть полностью загружены в память в структурах данных Python, таких как списки или словари, обеспечивая быстрый доступ за счет использования памяти. В качестве альтернативы файлы можно читать с диска по мере необходимости, с меньшей производительностью, но меньшими требованиями к памяти.
>>># Этот сценарий загружает файл, содержащий несколько последовательностей, и сохраняет каждую в другом формате.>>>from Bio import SeqIO>>>genomes = SeqIO.parse ('salmonella.gb', 'genbank')>>>для генома в геномах:... SeqIO.write (genome, genome.id + '.fasta ',' fasta ')С помощью модуля Bio.Entrez пользователи Biopython могут загружать биологические данные из баз данных NCBI. Каждая из функций, предоставляемых поисковой системой Entrez, доступна через функции в этом модуле, включая поиск и загрузку записей.
>>># Этот сценарий загружает геномы из базы данных NCBI Nucleotide и сохраняет их в файл FASTA.>>>из Bio import Entrez>>>из Bio import SeqIO>>>output_file = open ('all_records.fasta', "w")>>>Entrez.email = '[email#160;protected] '>>>records_to_download = ['FO834906.1', 'FO203501.1']>>>для record_id в records_to_download:... handle = Entrez.efetch (db = 'nucleotide', id = record_id, rettype = 'gb')... seqRecord = SeqIO.read (handle, format = 'gb')... handle.close ()... output_file.write (seqRecord.format ('fasta')) Рис. 1: Филогенетические корни дерево, созданное Bio.Phylo, показывающее взаимосвязь между гомологами Apaf-1 разных организмов
Рис. 1: Филогенетические корни дерево, созданное Bio.Phylo, показывающее взаимосвязь между гомологами Apaf-1 разных организмов 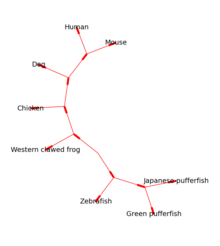 Рисунок 2: То же дерево, что и выше, нарисованное без корней с помощью Graphviz через Bio.Phylo
Рисунок 2: То же дерево, что и выше, нарисованное без корней с помощью Graphviz через Bio.Phylo Модуль Bio.Phylo предоставляет инструменты для работы с и визуализация филогенетических деревьев. Для чтения и записи поддерживаются различные форматы файлов, включая Newick, NEXUS и phyloXML. Общие манипуляции с деревом и обходы поддерживаются с помощью объектов Treeи Clade. Примеры включают преобразование и упорядочение файлов деревьев, извлечение подмножеств из дерева, изменение корня дерева и анализ характеристик ветвей, таких как длина или оценка.
Деревья с корнем могут быть нарисованы в ASCII или с использованием matplotlib (см. Рисунок 1) и библиотеку Graphviz можно использовать для создания некорневых макетов (см. Рисунок 2).
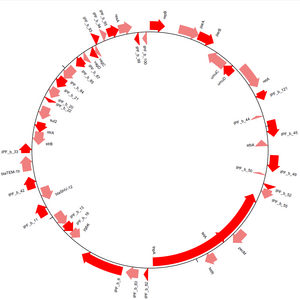 Рисунок 3: Диаграмма генов на плазмиде pKPS77, визуализированная с помощью модуля GenomeDiagram в Biopython
Рисунок 3: Диаграмма генов на плазмиде pKPS77, визуализированная с помощью модуля GenomeDiagram в Biopython Модуль GenomeDiagram предоставляет методы визуализации последовательностей в Biopython. Последовательности можно рисовать в линейной или круговой форме (см. Рисунок 3), и поддерживаются многие форматы вывода, включая PDF и PNG. Диаграммы создаются путем создания треков и последующего добавления к ним функций последовательности. Перебирая объекты последовательности и используя их атрибуты, чтобы решить, добавляются ли они к дорожкам диаграммы и каким образом, можно в значительной степени контролировать внешний вид окончательной диаграммы. Между разными дорожками можно провести перекрестные ссылки, что позволяет сравнивать несколько последовательностей на одной диаграмме.
Модуль Bio.PDB может загружать молекулярные структуры из файлов PDB и mmCIF и был добавлен в Biopython в 2003 году. Объект структурыявляется центральным для этого модуля, и он организует структуру макромолекул в иерархическом порядке: объекты Структурасодержат объекты Модель, которые содержат Цепьобъекты, которые содержат объекты Остаток, которые содержат объекты Atom. Неупорядоченные остатки и атомы получают свои собственные классы, DisorderedResidueи DisorderedAtom, которые описывают их неопределенные положения.
Используя Bio.PDB, можно перемещаться по отдельным компонентам файла макромолекулярной структуры, например исследовать каждый атом в белке. Могут быть выполнены общие анализы, такие как измерение расстояний или углов, сравнение остатков и расчет глубины остатков.
Модуль Bio.PopGen добавляет поддержку Biopython для Genepop, программного пакета для статистического анализа популяционной генетики. Это позволяет анализировать равновесие Харди – Вайнберга, неравновесие по сцеплению и другие характеристики популяции частот аллелей.
. Этот модуль также может выполнять популяционное генетическое моделирование с использованием объединяющая теория с программой fastsimcoal2.
Многие модули Biopython содержат оболочки командной строки для часто используемых инструментов, что позволяет использовать эти инструменты изнутри Biopython. Эти оболочки включают BLAST, Clustal, PhyML, EMBOSS и SAMtools. Пользователи могут создать подкласс общего класса оболочки, чтобы добавить поддержку любого другого инструмента командной строки.