
Войти
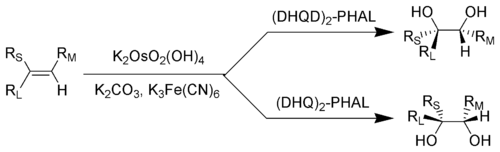 В реакции дигидроксилирования Шарплесса хиральность продукта можно контролировать с помощью «AD- смесь "использовалась. Это пример энантиоселективного синтеза с использованием асимметричной индукции.. Обозначение: R L = наибольший заместитель; R233>M8 = заместитель среднего размера; R S = наименьший заместитель
В реакции дигидроксилирования Шарплесса хиральность продукта можно контролировать с помощью «AD- смесь "использовалась. Это пример энантиоселективного синтеза с использованием асимметричной индукции.. Обозначение: R L = наибольший заместитель; R233>M8 = заместитель среднего размера; R S = наименьший заместитель 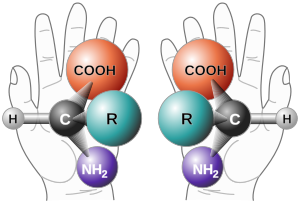 Два энантиомера общей альфа-аминокислоты
Два энантиомера общей альфа-аминокислоты Энантиоселективный синтез, также называемый асимметричным синтезом, представляет собой форму химического синтез. Он определяется в IUPAC как: химическая реакция (или последовательность реакций), в которой один или несколько новых элементов хиральности образуются в молекуле субстрата и которая дает стереоизомер (энантиомерные или диастереоизомерные ) продукты в неравных количествах.
Проще говоря: это синтез соединения методом, который способствует образованию специфический энантиомер или диастереомер. Энантиомеры - это стереоизомеры, которые имеют противоположные конфигурации в каждом хиральном центре. Диастереомеры - это стереоизомеры, которые различаются по одному или нескольким хиральным центрам.
Энантиоселективный синтез является ключевым процессом в современной химии и особенно важен в области фармацевтики, поскольку различные энантиомеры или диастереомеры молекулы часто имеют разную биологическую активность.
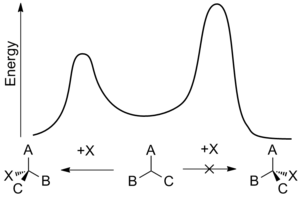 энергетический профиль энантиоселективной реакции присоединения.
энергетический профиль энантиоселективной реакции присоединения. Многие из строительных блоков биологических систем такие как сахара и аминокислоты, продуцируются исключительно в виде одного энантиомера. В результате живые системы обладают высокой степенью химической хиральности и часто будут по-разному реагировать с различными энантиомерами данного соединения. Примеры этой селективности включают:
Как таковой энантиоселективный синтез имеет большое значение, но может быть и труднодостижимым. Энантиомеры обладают идентичными энтальпиями и энтропиями и, следовательно, должны производиться в равных количествах с помощью ненаправленного процесса, приводящего к рацемической смеси. Энантиоселективный синтез может быть достигнут за счет использования хирального признака, который способствует образованию одного энантиомера по сравнению с другим посредством взаимодействий в переходном состоянии . Это смещение известно как асимметричная индукция и может включать хиральные особенности в субстрате, реагенте, катализаторе или окружающей среде и работает, создавая энергия активации, необходимая для образования одного энантиомера ниже, чем у противоположного энантиомера.
Энантиоселективность обычно определяется относительной скоростью стадии энантиодифференцирования - точки, в которой один реагент может стать либо двух энантиомерных продуктов. константа скорости, k, для реакции является функцией энергии активации реакции, иногда называемой энергетическим барьером, и зависит от температуры. Использование свободной энергии Гиббса энергетического барьера ΔG * означает, что относительные скорости противоположных стереохимических исходов при заданной температуре T равны:

Эта температурная зависимость означает, что разница скоростей и, следовательно, энантиоселективность больше при более низких температурах. В результате даже небольшие перепады энергетических барьеров могут дать заметный эффект.
| ΔΔG * (ккал) | k1/k2при 273 K | k1/k2при 298 K | k1/k2при 323 K) | |||
|---|---|---|---|---|---|---|
| 1,0 | 6. | 0,37 | 5. | 0,46 | 4. | 0,78 |
| 2,0 | 40. | .6 | 29. | .8 | 22. | .9 |
| 3,0 | 259 | 162 | 109 | |||
| 4,0 | 1650 | 886 | 524 | |||
| 5,0 | 10500 | 4830 | 2510 | |||
Энантиоселективный катализ (традиционно известный как асимметричный катализ) выполняется с хиральными катализаторами. Это либо биокатализаторы (например, ферменты), либо хиральные органокатализаторы, либо хиральные координационные комплексы. Катализ эффективен для более широкого круга превращений, чем любой другой метод энантиоселективного синтеза. Хиральные металлические катализаторы почти всегда становятся хиральными за счет использования хиральных лигандов (однако возможно получение хиральных комплексов с металлом, полностью состоящих из ахиральных лигандов, и такие хиральные по -металлические катализаторы недавно были продемонстрированы как очень полезные). Большинство энантиоселективных катализаторов эффективны при низких соотношениях субстрат / катализатор. Учитывая их высокую эффективность, они часто подходят для синтеза в промышленных масштабах даже с использованием дорогих катализаторов. Универсальным примером энантиоселективного синтеза является асимметричное гидрирование, которое используется для восстановления широкого спектра функциональных групп.

При разработке новых катализаторов в значительной степени доминирует разработка новых классов лиганды. Определенные лиганды, часто называемые «привилегированными лигандами », оказались эффективными в широком диапазоне реакций; примеры включают BINOL, Salen и BOX. Однако в целом несколько катализаторов эффективны более чем в одном типе асимметричной реакции. Например, для асимметричного гидрирования Нойори с помощью BINAP / Ru требуется β-кетон, хотя другой катализатор, BINAP / диамин-Ru, расширяет область действия до α, β- алкенов и <290.>ароматические химические вещества.
Хиральные вспомогательные вещества - это органическое соединение, которое соединяется с исходным материалом с образованием нового соединения, которое затем может вступать в энантиоселективные реакции посредством внутримолекулярной асимметричной индукции. По окончании реакции вспомогательное вещество удаляют в условиях, которые не вызывают рацемизации продукта. Затем он обычно восстанавливается для использования в будущем.

Хиральные вспомогательные вещества должны использоваться в стехиометрических количествах, чтобы быть эффективными и требовать дополнительных стадий синтеза для добавления и удаления вспомогательного вещества. Однако в некоторых случаях единственная доступная стереоселективная методология полагается на хиральные вспомогательные вещества, и эти реакции имеют тенденцию быть универсальными и очень хорошо изученными, что обеспечивает наиболее эффективный по времени доступ к энантиомерно чистым продуктам. Кроме того, продуктами реакций, направленных на вспомогательные вещества, являются диастереомеры, что делает возможным их легкое разделение такими методами, как колоночная хроматография или кристаллизация.
Биокатализ использует биологические соединения, от изолированных ферментов до живых клеток, для проведения химических превращений. Преимущества этих реагентов включают очень высокую e.e.s и специфичность реагента, а также мягкие рабочие условия и низкое воздействие на окружающую среду. Биокатализаторы чаще используются в промышленности, чем в академических исследованиях; например, при производстве статинов. Однако высокая специфичность реагента может быть проблемой, поскольку часто требуется, чтобы перед нахождением эффективного реагента был проведен скрининг широкого спектра биокатализаторов.
Органокатализ относится к форме катализа, при котором скорость химической реакции увеличивается за счет органического соединения состоящий из углерода, водорода, серы и других неметаллических элементов. Когда органокатализатор хиральный, тогда может быть достигнут энантиоселективный синтез; например, ряд реакций образования углерод-углеродной связи становится энантиоселективным в присутствии пролина, причем альдольная реакция является ярким примером. В органокатализе в качестве хиральных катализаторов часто используются природные соединения и вторичные амины ; они недорогие и экологически чистые, поскольку в них не используются металлы.
Синтез хирального пула - один из самых простых и старых подходов к энантиоселективному синтезу. Легкодоступным хиральным исходным материалом манипулируют посредством последовательных реакций, часто с использованием ахиральных реагентов, для получения желаемой целевой молекулы. Это может соответствовать критериям энантиоселективного синтеза, когда создается новый хиральный вид, например, в реакции SN2..

Синтез хирального пула особенно привлекателен для молекул-мишеней, имеющих хиральность, подобную относительно недорогим естественным строительным блокам, таким как в виде сахара или аминокислоты. Однако количество возможных реакций, которым может подвергаться молекула, ограничено, и могут потребоваться сложные пути синтеза (например, общий синтез осельтамивира ). Этот подход также требует стехиометрического количества энантиочистки исходного материала, что может быть дорогостоящим, если не встречается в природе.
Два энантиомера молекулы обладают одинаковыми физическими свойствами (например, точка плавления, точка кипения, полярность и т. д.) и поэтому ведут себя идентично друг другу. В результате они будут мигрировать с идентичным R f в тонкослойной хроматографии и будут иметь идентичные времена удерживания в HPLC и GC. Их спектры ЯМР и ИК идентичны.
Это может затруднить определение того, дает ли процесс единственный энантиомер (и, что особенно важно, какой это энантиомер), а также затруднить отделение энантиомеров от реакции, которая не была на 100% энантиоселективной. К счастью, энантиомеры по-разному ведут себя в присутствии других хиральных материалов, и это можно использовать для их разделения и анализа.
Энантиомеры не мигрируют одинаково на хиральных хроматографических средах, таких как кварц или стандартных средах, которые были хирально модифицированы. Это составляет основу хиральной колоночной хроматографии, которую можно использовать в небольшом масштабе для проведения анализа с помощью ГХ и ВЭЖХ или в большом масштабе для разделения хирально нечистые материалы. Однако для этого процесса может потребоваться большое количество хирального упаковочного материала, что может быть дорогостоящим. Распространенной альтернативой является использование хирального дериватизирующего агента для превращения энантиомеров в диастереомеры во многом таким же образом, как хиральные вспомогательные вещества. Они имеют разные физические свойства и, следовательно, могут быть разделены и проанализированы с использованием обычных методов. В ЯМР-спектроскопии стереоизомеров используются специальные хиральные дериватизирующие агенты, известные как «агенты хирального разделения», обычно они включают координацию с хиральными комплексами европия, такими как Eu (fod) 3 и Eu (hfc) 3.
. Энантиомерный избыток вещества также можно определить с помощью определенных оптических методов. Самый старый способ сделать это - использовать поляриметр для сравнения уровня оптического вращения в продукте со «стандартом» известного состава. Также возможно выполнить ультрафиолетовую и видимую спектроскопию стереоизомеров, используя эффект Коттона.
. Один из наиболее точных способов определения хиральности соединения - определение его абсолютной конфигурации. от Рентгеновская кристаллография. Однако это трудоемкий процесс, который требует выращивания подходящего монокристалла.
В 1815 году французский физик Жан-Батист Био показал, что некоторые химические вещества могут вращать плоскость луч поляризованного света, свойство, называемое оптической активностью. Природа этого свойства оставалась загадкой до 1848 года, когда Луи Пастер предположил, что у него есть молекулярная основа, происходящая из некоторой формы «диссимметрии», а термин хиральность был придуман лордом Кельвином Год спустя. Происхождение самой хиральности было окончательно описано в 1874 году, когда Якобус Хенрикус ван 'т Хофф и Джозеф Ле Бель независимо друг от друга предложили тетраэдрическую геометрию углерода. Структурные модели до этой работы были двумерными, и Ван'т Хофф и Ле Бель предположили, что расположение групп вокруг этого тетраэдра может определять оптическую активность полученного соединения посредством того, что стало известно как Ле Бель– Правило Ван 'т Гоффа.
 Энантиоселективное декарбоксилирование 2-этил-2-метил малоновой кислоты, катализируемое Марквальдом бруцином, в результате чего образуется небольшой избыток левовращающей формы 2- продукт метилмасляной кислоты.
Энантиоселективное декарбоксилирование 2-этил-2-метил малоновой кислоты, катализируемое Марквальдом бруцином, в результате чего образуется небольшой избыток левовращающей формы 2- продукт метилмасляной кислоты. В 1894 году Герман Эмиль Фишер изложил концепцию асимметричной индукции ; в котором он правильно приписал селективное образование D -глюкозы растениями за счет влияния оптически активных веществ в хлорофилле. Фишер также успешно выполнил то, что теперь считается первым примером энантиоселективного синтеза, путем энантиоселективного удлинения сахаров с помощью процесса, который в конечном итоге стал синтезом Килиани-Фишера.
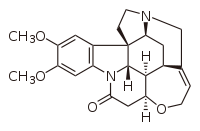 Бруцин, алкалоид натуральный продукт, относящийся к стрихнину, успешно использованный в качестве органокатализатора Марквальдом в 1904 году.
Бруцин, алкалоид натуральный продукт, относящийся к стрихнину, успешно использованный в качестве органокатализатора Марквальдом в 1904 году. Первый энантиоселективный химический синтез чаще всего приписывается Вилли Marckwald, Universität zu Berlin, для катализируемого бруцином энантиоселективного декарбоксилирования 2-этил-2-метил малоновой кислоты сообщалось в 1904 году. Был образован небольшой избыток левовращающей формы продукта реакции, 2-метилмасляной кислоты; поскольку этот продукт также является натуральным продуктом, например, в виде боковой цепи ловастатина, образованной его дикетидсинтазой (LovF) во время его биосинтеза, - этот результат представляет собой первый зарегистрированный полный синтез с энантиоселективностью, а также другие первые (как отмечает Коскинен, первый «пример асимметричного катализа, энантиотопического отбора и органокатализа »). Это наблюдение также имеет историческое значение, поскольку в то время энантиоселективный синтез можно было понять только в терминах витализма. В то время многие выдающиеся химики, такие как Йенс Якоб Берцелиус, утверждали, что естественные и искусственные соединения фундаментально отличаются и что хиральность была просто проявлением «жизненной силы», которая могла существовать только в природных соединениях. В отличие от Фишера, Марквальд провел энантиоселективную реакцию с ахиральным, неприродным исходным материалом, хотя и с хиральным органокатализатором (как мы теперь понимаем эту химию).
Развитие энантиоселективного синтеза изначально шло медленно, в основном из-за ограниченного набора доступных методов их разделения и анализа. Диастереомеры обладают разными физическими свойствами, что позволяет разделить их обычными способами, однако в то время энантиомеры можно было разделить только с помощью спонтанного разделения (где энантиомеры разделяются при кристаллизации) или кинетического разрешения (где один энантиомер избирательно уничтожается). Единственным инструментом для анализа энантиомеров была оптическая активность с использованием поляриметра, метод, который не предоставляет структурных данных.
Настоящий прогресс начался только в 1950-х годах. Частично движимый химиками, такими как Р. Б. Вудворд и Владимир Прелог, но также путем разработки новых методов. Первым из них была рентгеновская кристаллография, которая была использована для определения абсолютной конфигурации органического соединения Йоханнесом Бийвоет в 1951 году. Была введена хиральная хроматография. годом позже Дэлглишем, который использовал бумажную хроматографию для разделения хиральных аминокислот. Хотя Дэлглиш не был первым, кто наблюдал такое разделение, он правильно приписал разделение энантиомеров различному удерживанию хиральной целлюлозой. Это было расширено в 1960 году, когда Клем и Рид впервые сообщили об использовании хирально-модифицированного силикагеля для хирального ВЭЖХ разделения.
 Два энантиомера талидомида:. Слева: (S) - талидомид. Справа: (R) -талидомид
Два энантиомера талидомида:. Слева: (S) - талидомид. Справа: (R) -талидомид Хотя было известно, что разные энантиомеры лекарства могут иметь разную активность, при этом значительную раннюю работу проделал Артур Робертсон Кушни, это не было учтено при разработке и тестировании первых лекарств. Однако после катастрофы с талидомидом разработка и лицензирование лекарств резко изменились.
Впервые синтезированный в 1953 году, талидомид широко применялся при утреннем недомогании с 1957 по 1962 год, но вскоре было обнаружено, что он обладает серьезным тератогенным действием, в конечном итоге вызывая врожденные дефекты у более чем 10 000 младенцев. Катастрофа побудила многие страны ввести более жесткие правила тестирования и лицензирования лекарств, такие как Поправка Кефовера-Харриса (США) и Директива 65/65 / EEC1 (ЕС).
Ранние исследования тератогенного механизма с использованием мышей показали, что один энантиомер талидомида обладает тератогенным действием, тогда как другой обладает всей терапевтической активностью. Позже было показано, что эта теория неверна, и теперь ее опровергли исследования. Однако это повысило важность хиральности при разработке лекарств, что привело к расширению исследований в области энантиоселективного синтеза.
Правила приоритета Кана – Ингольда – Прелога (часто сокращенно обозначаемые как система CIP ) были впервые опубликованы в 1966 году; позволяя более легко и точно описывать энантиомеры. В том же году произошло первое успешное разделение энантиомеров с помощью газовой хроматографии, что стало важным достижением, поскольку в то время технология была широко распространена.
Энантиоселективный синтез, катализируемый металлами, впервые был предложен Уильямом С. Ноулзом, Рёдзи Нойори и К. Барри Шарплесс ; за что они получили бы 2001 Нобелевскую премию по химии. Ноулз и Нойори начали с разработки асимметричного гидрирования, которое они разработали независимо в 1968 году. Ноулз заменил ахиральные трифенилфосфин лиганды в катализаторе Уилкинсона на хиральный фосфиновые лиганды. Этот экспериментальный катализатор использовали в асимметричном гидрировании с умеренным 15% энантиомерным избытком. Ноулз был также первым, кто применил энантиоселективный металлический катализ для синтеза в промышленных масштабах; работая в Monsanto Company, он разработал стадию энантиоселективного гидрирования для производства L-DOPA с использованием лиганда DIPAMP.
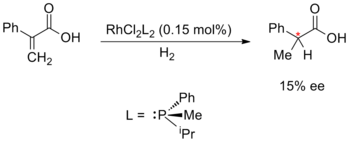 |  | |
| Ноулз: Асимметричное гидрирование (1968) | Нойори: энантиоселективное циклопропанирование (1968) |
|---|
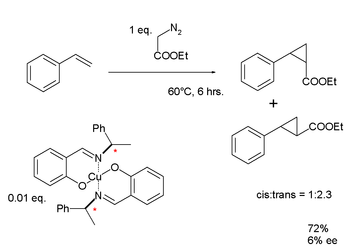
Нойори разработал комплекс меди с использованием хирального лиганда основания Шиффа, который он использовал для циклопропанирования металл-карбеноид стирол. Как и результаты Ноулза, результаты Нойори для энантиомерного избытка для этого лиганда первого поколения были разочаровывающе низкими: 6%. Однако продолжающиеся исследования в конечном итоге привели к разработке реакции асимметричного гидрирования Нойори.
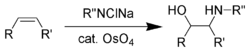 Оксиаминирование Шарплесса
Оксиаминирование Шарплесса Шарплесс дополнил эти реакции восстановления, разработав ряд асимметричных окислений (эпоксидирование Шарплесса, асимметричное дигидроксилирование Шарплесса, оксиаминирование Шарплесса ) в течение 1970-х и 1980-х годов. Самой ранней из них является реакция асимметричного оксиаминирования с использованием четырехокиси осмия.
В тот же период были разработаны методы, позволяющие анализировать хиральные соединения с помощью ЯМР ; либо с использованием хиральных дериватизирующих агентов, таких как кислота Мошера, либо сдвигающих реагентов на основе европия, из которых Eu (DPM) 3 был самым ранним.
Хиральные вспомогательные вещества были представлены EJ Кори в 1978 году и занимал видное место в творчестве Дитера Эндерса. Примерно в то же время был разработан энантиоселективный органокатализ с новаторскими работами, включая реакцию Хайоса-Пэрриша-Эдера-Зауэра-Вихерта. Энантиоселективные реакции, катализируемые ферментами, становились все более и более распространенными в течение 1980-х годов, особенно в промышленности, с их применением, включая гидролиз асимметричного сложного эфира с помощью эстеразы печени свиньи. Новые технологии генной инженерии позволили адаптировать ферменты к конкретным процессам, допуская расширенный диапазон селективных преобразований. Например, в асимметричном гидрировании предшественников статина.