
Войти
| Альдольная реакция | |
|---|---|
| Тип реакции | Реакция связывания |
| Идентификаторы | |
| Портал органической химии | альдол- дополнение |
| RSC ID онтологии | RXNO: 0000016 |
альдольная реакция является средством образования углеродно-углеродных связей в органической химии. Эта реакция, открытая независимо русским химиком Александром Бородиным в 1869 году и французским химиком Шарлем-Адольфом Вюрцем в 1872 году, объединяет два карбонильных соединения (первоначальные эксперименты использовали альдегиды ) для образования нового β-гидроксикарбонильного соединения. Эти продукты известны как альдолы, из альдегида + спирта, структурного мотива, наблюдаемого во многих продуктах. Структурные единицы альдола встречаются во многих важных молекулах, как природных, так и синтетических. Например, альдольная реакция использовалась в крупномасштабном производстве товарного химического вещества пентаэритрит и в синтезе лекарственного средства от сердечных заболеваний липитор (аторвастатин, кальциевая соль).
Альдольная реакция объединяет две относительно простые молекулы в более сложную. Повышенная сложность возникает из-за образования до двух новых стереогенных центров (на α- и β-углероде альдольного аддукта, отмеченных звездочками на схеме ниже). Современная методология позволяет не только позволить альдольным реакциям протекать с высоким выходом, но также управлять как относительной, так и абсолютной конфигурацией этих стереоцентров. Эта способность избирательно синтезировать конкретный стереоизомер важна, потому что разные стереоизомеры могут иметь очень разные химические и биологические свойства.
Например, стереогенные альдольные звенья особенно распространены в поликетидах, классе молекул, обнаруживаемых в биологических организмах. В природе поликетиды синтезируются ферментами, которые вызывают итерационные конденсации Клайзена. 1,3-дикарбонильные продукты этих реакций затем могут быть подвергнуты различным дериватизам с образованием широкого разнообразия интересных структур. Часто такая дериватизация включает восстановление одной из карбонильных групп с образованием альдольной субъединицы. Некоторые из этих структур обладают мощными биологическими свойствами: иммунодепрессант FK506, противоопухолевый агент дискодермолид или противогрибковое агент амфотерицин B, например. Хотя синтез многих таких соединений когда-то считался почти невозможным, альдольная методология позволила во многих случаях их эффективный синтез.
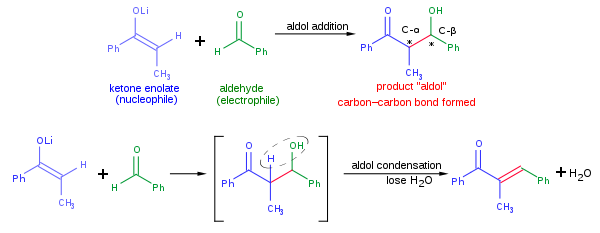
Типичная современная реакция присоединения альдола , показанная выше, может включают нуклеофильное присоединение енолята кетона к альдегиду. После образования альдольный продукт может иногда терять молекулу воды с образованием α, β-ненасыщенного карбонильного соединения. Это называется альдольной конденсацией. В альдольной реакции можно использовать различные нуклеофилы, включая енолы, енолаты и енольные простые эфиры кетонов, альдегидов и многие другие карбонильные соединения. Электрофильный партнер обычно представляет собой альдегид или кетон (существует множество вариантов, таких как реакция Манниха ). Когда нуклеофил и электрофил различны, реакция называется перекрестной альдольной реакцией; наоборот, когда нуклеофил и электрофил совпадают, реакция называется альдольной димеризацией.
 Типичная экспериментальная установка для альдольной реакции.. Колба справа представляет собой раствор диизопропиламид лития (LDA) в тетрагидрофуране (THF). Колба слева представляет собой раствор енолята лития и трет-бутилпропионата (образованного добавлением LDA к трет-бутилпропионату). Затем в енолатную колбу можно добавить альдегид, чтобы инициировать реакцию присоединения альдола.. Обе колбы погружают в охлаждающую баню сухой лед / ацетон (-78 ° C), температура которой контролируется термопарой (провод слева).
Типичная экспериментальная установка для альдольной реакции.. Колба справа представляет собой раствор диизопропиламид лития (LDA) в тетрагидрофуране (THF). Колба слева представляет собой раствор енолята лития и трет-бутилпропионата (образованного добавлением LDA к трет-бутилпропионату). Затем в енолатную колбу можно добавить альдегид, чтобы инициировать реакцию присоединения альдола.. Обе колбы погружают в охлаждающую баню сухой лед / ацетон (-78 ° C), температура которой контролируется термопарой (провод слева). Альдольные реакции могут протекать по двум принципиально различным механизмам. Карбонильные соединения, такие как альдегиды и кетоны, можно превратить в енолы или простые енольные эфиры. Эти частицы, будучи нуклеофильными по α-углероду, могут атаковать особенно реакционноспособные протонированные карбонилы, такие как протонированные альдегиды. Это «енольный механизм». Карбонильные соединения, являющиеся углеродными кислотами, также могут быть депротонированы с образованием енолятов, которые являются гораздо более нуклеофильными, чем енолы или енольные эфиры, и могут напрямую атаковать электрофилы. Обычный электрофил - это альдегид, поскольку кетоны гораздо менее реакционноспособны. Это «енолятный механизм».
Если условия особенно суровые (например: NaOMe / MeOH / орошение ), может произойти конденсация, но этого обычно можно избежать с помощью мягких реагентов и низких температур (например, LDA (a сильное основание), ТГФ, −78 ° C). Хотя добавление альдола обычно протекает почти до завершения в необратимых условиях, изолированные альдольные аддукты чувствительны к индуцированному основанием расщеплению ретроальдола с возвращением исходных материалов. Напротив, ретроальдольные конденсации редки, но возможны.

Когда используется кислотный катализатор, начальная стадия в механизме реакции включает катализируемый кислотой таутомеризация карбонильного соединения до енола. Кислота также служит для активации карбонильной группы другой молекулы путем протонирования, что делает ее высоко электрофильной. Енол является нуклеофильным по α-углероду, что позволяет ему атаковать протонированное карбонильное соединение, что приводит к альдолу после депротонирования. Обычно он дегидратируется с образованием ненасыщенного карбонильного соединения. На схеме показана типичная катализируемая кислотой самоконденсация альдегида.
Катализируемый кислотой альдольный механизм

Катализируемая кислотой дегидратация

Если катализатор представляет собой умеренное основание, такое как ион гидроксида или алкоксидом, альдольная реакция происходит посредством нуклеофильной атаки резонансно-стабилизированным енолятом на карбонильную группу другой молекулы. Продукт представляет собой алкоксид соль альдольного продукта. Затем образуется сам альдол, который затем может подвергаться дегидратации с образованием ненасыщенного карбонильного соединения. На схеме показан простой механизм альдольной реакции альдегида с самим собой, катализируемой основанием.
Катализируемая основанием альдольная реакция (показана с использованием OCH 3 в качестве основания)

Катализируемая основанием дегидратация (часто неправильно записывается как одностадийная, см. реакция элиминирования E1cB )

Хотя в некоторых случаях требуется только каталитическое количество основания, более обычной процедурой является использование стехиометрического количества сильного основания, такого как LDA или NaHMDS.. В этом случае образование енолята необратимо, и альдольный продукт не образуется до тех пор, пока алкоксид металла альдольного продукта не протонируется на отдельной стадии обработки.
Известны более совершенные формы механизма. В 1957 году Говард Циммерман и М.Д. Тракслер предположили, что некоторые альдольные реакции имеют «шестичленные переходные состояния, имеющие конформацию кресло ». теперь известна как модель Циммермана-Тракслера . Е-еноляты дают антипродукты, тогда как Z-еноляты дают син-продукты. Факторы, которыеСелективность контроля - это предпочтение экваториального размещения заместителей в шестичленных переходных состояниях и предотвращение син-пентановых взаимодействий соответственно. E и Z относятся к цис-транс стереохимическая связь между енолятом кислорода, несущим положительный противоион, и группой наивысшего приоритета на альфа-углероде. В действительности только некоторые металлы, такие как литий, достоверно следуют модели Циммермана – Тракслера. Таким образом, в некоторых случаях стереохимический исход реакции может быть непредсказуемым.

Проблема «контроля» при добавлении альдола лучше всего демонстрируется на примере. Рассмотрим результат этой гипотетической реакции:

В этой реакции два несимметричных кетона конденсируются с использованием этилата натрия. Основность этоксида натрия такова, что он не может полностью депротонировать ни один из кетонов, но может производить небольшие количества енолята натрия обоих кетонов. Это означает, что, помимо того, что оба кетона являются потенциальными альдольными электрофилами, они также могут действовать как нуклеофилы через свой енолят натрия. Таким образом, два электрофила и два нуклеофила потенциально могут дать четыре возможных продукта:

Таким образом, если кто-то хочет получить только один из перекрестных продуктов, нужно контролировать, какой карбонил становится нуклеофильным енолом / енолятом, а какой остается в его электрофильная карбонильная форма.
Простейший контроль - это если только один из реагентов имеет кислотные протоны, и только эта молекула образует енолят. Например, добавление диэтилмалоната к бензальдегиду приведет к получению только одного продукта. Только малонат имеет α-атомы водорода, поэтому он является нуклеофильным партнером, тогда как неэнолизируемый бензальдегид может быть только электрофилом:

Малонат особенно легко депротонировать, поскольку α-положение фланкируется более чем одним карбонилом. Двойная активация делает енолят более стабильным, поэтому для его образования требуется не такое сильное основание. Расширение этого эффекта может позволить контролировать, какой из двух карбонильных реагентов становится енолятом, даже если оба имеют α-атомы водорода. Если один партнер значительно более кислый, чем другой, наиболее кислый протон отщепляется основанием, и у этого карбонила образуется енолят, в то время как менее кислый карбонил не подвергается воздействию основания. Этот тип контроля работает только в том случае, если разница в кислотности достаточно велика и для реакции не используется избыток основания. Типичный субстрат для этой ситуации - это когда депротонируемое положение активируется более чем одной карбонилподобной группой. Общие примеры включают группу CH 2, фланкированную двумя карбонилами или нитрилами (см., Например, конденсацию Кневенагеля и первые стадии синтеза эфира малоновой кислоты ).
Одно из распространенных решений - сначала сформировать енолят одного партнера, а затем добавить другого партнера под кинетическим контролем. Кинетический контроль означает, что прямая реакция альдольного присоединения должна быть значительно быстрее, чем обратная ретро-альдольная реакция. Чтобы этот подход был успешным, также должны быть выполнены два других условия; должно быть возможно количественно образовать енолят одного партнера, и прямая альдольная реакция должна быть значительно быстрее, чем перенос енолят от одного партнера к другому. Общие условия кинетического контроля включают образование енолята кетона с LDA при -78 ° C с последующим медленным добавлением альдегида.
Енолят можно получить с использованием сильного основания («жесткие условия») или с использованием кислоты Льюиса и слабого base («мягкие условия»):
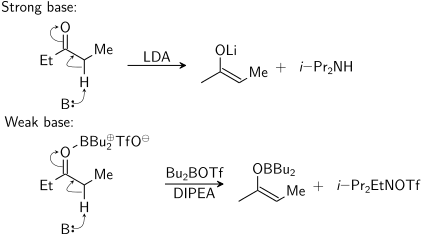
На этой диаграмме B: представляет собой основание, которое принимает протон. трифлат дибутилбора фактически присоединяется к кислороду только во время реакции. Второй продукт справа (образованный из N, N-диизопропилэтиламина ) должен быть i-Pr 2 EtNH OTf.
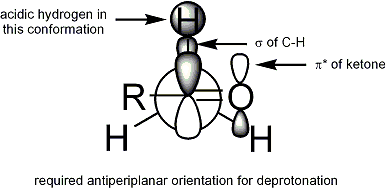
Для депротонирования стереоэлектронное требование состоит в том, чтобы сигма-связь альфа-CH могла перекрываться с pi * -орбиталью карбонила. :
Были проведены обширные исследования образования енолятов в различных условиях. В настоящее время возможно получение в большинстве случаев желаемой геометрии енолята:
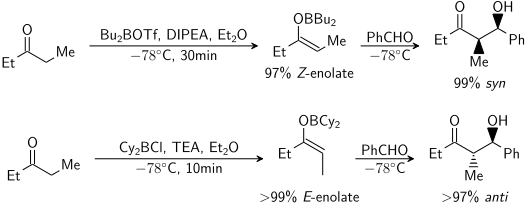
Для кетонов большинство условий енолизации дает Z еноляты. Для сложных эфиров большинство условий енолизации дают еноляты E. Известно, что добавление HMPA изменяет стереоселективность депротонирования.

Стереоселективное образование енолятов было рационализировано с помощью Ирландской модели , хотя ее достоверность несколько сомнительна. В большинстве случаев неизвестно, какие промежуточные соединения являются мономерными или олигомерными, если таковые имеются; тем не менее, модель Ирландии остается полезным инструментом для понимания енолятов.
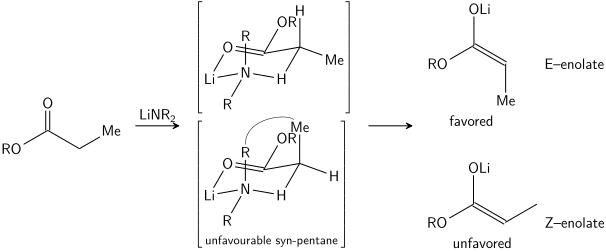
В модели Ирландии предполагается, что депротонирование происходит посредством шестичленного или циклического переходного состояния мономера. Более крупный из двух заместителей у электрофила (в приведенном выше случае метил больше, чем протон) занимает экваториальное расположение в предпочтительном переходном состоянии, что приводит к предпочтению енолятов E. Модель явно не работает во многих случаях; например, если смесь растворителей изменяется с THF на 23% HMPA-THF (как показано выше), геометрия енолята меняется на обратную, что несовместимо с этой моделью и его циклическим переходным состоянием.
Если несимметричный кетон подвергается действию основания, он может образовывать два региоизомерных енолята (без учета геометрии енолята). Например:
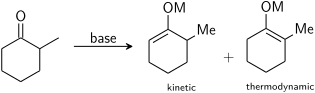
Тризамещенный енолят считается кинетическим енолятом, а тетразамещенный енолят считается термодинамическим енолятом. Альфа-водород, депротонированный с образованием кинетического енолята, менее затруднен и, следовательно, депротонируется быстрее. В общем, тетразамещенные олефины более стабильны, чем тризамещенные олефины, из-за гиперконъюгативной стабилизации. На соотношение енолятных региоизомеров сильно влияет выбор основания. Для приведенного выше примера кинетический контроль может быть установлен с помощью LDA при -78 ° C, что дает селективность кинетики: термодинамический енолят 99: 1, тогда как термодинамический контроль может быть установлен с помощью трифенилметиллития при комнатной температуре, что дает избирательность 10:90.
В целом кинетическим енолятам благоприятствуют низкие температуры, условия, которые обеспечивают относительно ионную связь металл-кислород, и быстрое депротонирование с использованием небольшого избытка сильного, стерически затрудненного основания. Большое основание депротонирует только более доступный водород, а низкие температуры и избыток основания помогают избежать уравновешивания с более стабильным альтернативным енолятом после начального образования енолята. Термодинамическим енолятам способствуют более длительное время уравновешивания при более высоких температурах, условия, которые дают относительно ковалентные связи металл-кислород, и использование небольшого субстехиометрического количества сильного основания. При использовании недостаточного количества основания для депротонирования всех молекул карбонила еноляты и карбонилы могут обмениваться протонами друг с другом и уравновешиваться со своим более стабильным изомером. Использование различных металлов и растворителей может обеспечить контроль над степенью ионности связи металл-кислород.
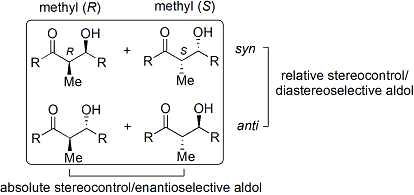
Альдольная реакция особенно полезна, поскольку в одной реакции образуются два новых стереогенных центра. Были проведены обширные исследования, чтобы понять механизм реакции и улучшить селективность, наблюдаемую во многих различных условиях. Условные обозначения син / анти обычно используются для обозначения относительной стереохимии α- и β-углерода.
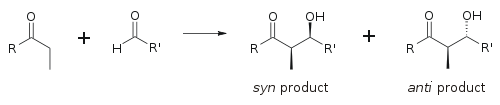
Это соглашение применяется, когда к альдегидам добавляются нуклеофилы пропионата (или более высокого порядка). Группа R кетона и группа R 'альдегида выровнены в виде «зигзагообразного» рисунка в плоскости бумаги (или экрана), и расположение сформированных стереоцентров считается синонимом или анти, в зависимости от того, находятся на одной или противоположных сторонах основной цепи.
В более старых работах используется номенклатура эритро / трео, знакомая по химии сахаридов.
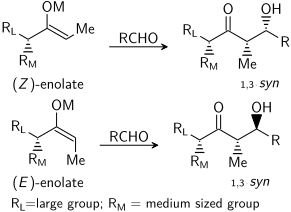
Не существует существенной разницы между уровнем стереоиндукции, наблюдаемой для енолятов E и Z. Каждая геометрия алкена приводит в первую очередь к одной определенной относительной стереохимии в продукте, E дает анти, а Z дает син:


Катион енолятного металла может играть большую роль в определении уровня стереоселективности в альдольная реакция. Бор часто используется, потому что его длина связи значительно короче, чем у металлов, таких как литий, алюминий или магний..

Например, связи бор – углерод и бор – кислород имеют длину 1,4–1,5 Å и 1,5–1,6 Å соответственно, тогда как типичные связи металл-углерод и металл-кислород составляют 1,9–2,2 Å. и 2,0–2,2 Å соответственно. Использование бора, а не металла «сжимает» переходное состояние и дает большую стереоселективность реакции. Таким образом, указанная выше реакция дает соотношение син: анти, равное 80:20 при использовании енолята лития по сравнению с 97: 3 при использовании енолята бибутилбора.
Альдольная реакция может проявлять «стереоконтроль на основе субстрата», в котором существующая хиральность любого реагента влияет на стереохимический результат реакции. Это было тщательно изучено, и во многих случаях можно предсказать смысл асимметричной индукции, если не абсолютный уровень диастереоселективности. Если енолят содержит стереоцентр в альфа-позиции, может быть реализован отличный стереоконтроль.
В случае енолята E доминирующим элементом управления является аллильный 1,3-штамм, тогда как в случае енолята Z доминирующим элементом управления является предотвращение 1,3-диаксиального взаимодействия. Общая модель представлена ниже:

Для ясности стереоцентр на енолате был эпимеризован ; в действительности, атака была бы направлена на противоположную диастереофасаду альдегида. В обоих случаях предпочтение отдается диастереомеру 1,3-син. Есть много примеров такого типа стереоконтроля:

Когда енолаты атакуют альдегиды с альфа-стереоцентром, также возможно отличное стереоконтроль. Общее наблюдение состоит в том, что еноляты Е проявляют диастереофазную селекцию Фелкина, тогда как енолиты Z проявляют селективность против Фелкина. Общая модель представлена ниже:
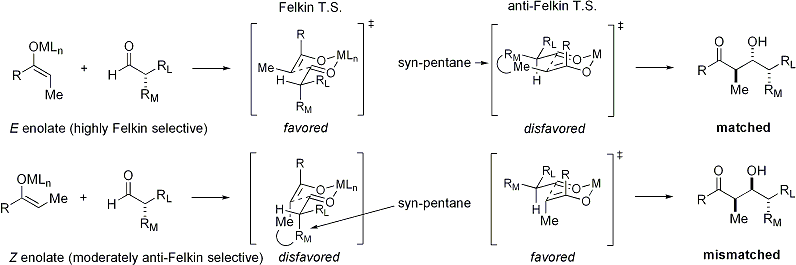
Поскольку Z-еноляты должны реагировать через переходное состояние, которое содержит либо дестабилизирующее син-пентановое взаимодействие, либо анти-Фелкин ротамер, Z-еноляты в этом случае проявляют более низкие уровни диастереоселективности. Некоторые примеры представлены ниже:

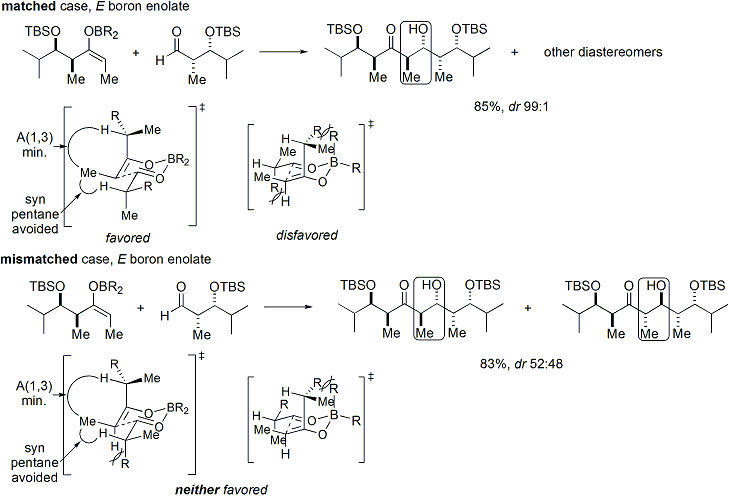
Если и енолят, и альдегид содержат ранее существовавшую хиральность, то результат альдольной реакции «двойной стереодифференциации» можно предсказать, используя объединенный стереохимический анализ. модель, которая учитывает смещение лица енолята, геометрию енолята и смещение лица альдегида. Несколько примеров применения этой модели приведены ниже:
Современные органические синтезы теперь требуют синтеза соединений в энантиочистой форме. Поскольку реакция присоединения альдола создает два новых стереоцентра, может получиться до четырех стереоизомеров.
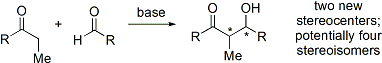
Было разработано множество методов, которые контролируют как относительную стереохимию (т.е. син- или анти-, как обсуждалось выше), так и абсолютную стереохимию (т.е. R или S).
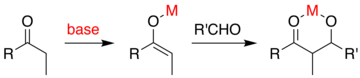
Широко используемым методом является метод ацил оксазолидинона Эванса. Разработанный в конце 1970-х и 1980-х годах Дэвидом А. Эвансом и сотрудниками, этот метод работает путем временного создания хирального енолята путем добавления хирального вспомогательного вещества. Ранее существовавшая хиральность от вспомогательного вещества затем переносится на альдольный аддукт путем проведения диастереоселективной альдольной реакции. При последующем удалении вспомогательного вещества обнаруживается желаемый стереоизомер альдола.
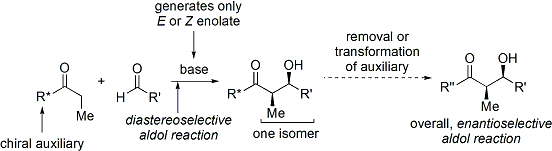
В случае метода Эванса добавляемым хиральным вспомогательным веществом является оксазолидинон, а полученное карбонильное соединение представляет собой имид. Ряд оксазолидинонов в настоящее время легко доступен в обеих энантиомерных формах. Они могут стоить примерно 10–20 долларов за грамм, что делает их относительно дорогими. Однако энантиочистые оксазолидиноны получают в 2 стадии синтеза из сравнительно недорогих аминокислот, а это означает, что крупномасштабный синтез можно сделать более экономичным за счет собственного приготовления. Обычно это включает опосредованное борогидридом восстановление кислотной части с последующей конденсацией / циклизацией полученного аминоспирта с простым карбонатным эфиром, таким как диэтилкарбонат.
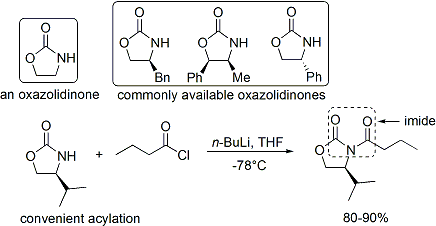
ацилирование оксазолидинона является удобной процедурой и неофициально называется «завершенная загрузка». Z-еноляты, приводящие к син-альдольным аддуктам, могут быть надежно образованы с использованием опосредованной бором мягкой енолизации:

Часто единственный диастереомер может быть получен путем одной кристаллизации альдольный аддукт. Однако с помощью метода Эванса нельзя надежно получить антиальдольные аддукты. Несмотря на стоимость и ограничение использования только синаддуктов, превосходная надежность, простота использования и универсальность метода делают его предпочтительным во многих ситуациях. Для расщепления вспомогательного вещества доступно множество методов:

После конструирования имида могут быть выполнены как син-, так и антиселективные реакции присоединения альдола, что позволяет собрать три из четырех возможных стереомассивов: синселективный: и антиселективный. селективный:

В син-селективных реакциях оба метода енолизации дают енолят Z, как и ожидалось; однако стереохимический результат реакции контролируется метилстереоцентром, а не хиральностью оксазолидинона. Описанные методы позволяют стереоселективную сборку поликетидов, класса натуральных продуктов, которые часто содержат альдольный ретрон.
 Рис. 1: Механизм типичной внутримолекулярной альдольной реакции в основных условиях.
Рис. 1: Механизм типичной внутримолекулярной альдольной реакции в основных условиях. Внутримолекулярная альдольная реакция представляет собой реакцию конденсации двух альдегидных групп или кетонных групп в одной и той же молекуле. Пяти- или шестичленные α, β-ненасыщенные кетон или альдегиды образуются в виде продуктов. Эта реакция является важным подходом к образованию углерод-углеродных связей в органических молекулах, содержащих кольцевые системы. Например, в сильнощелочных условиях (например, гидроксид натрия ), гексан-2,5-дион (соединение A на Фиг.1) может циклизоваться посредством внутримолекулярной альдольной реакции с образованием 3 -метилциклопент-2-ен-1-он (соединение B).
Механизм внутримолекулярной альдольной реакции включает образование ключевого енолятного промежуточного соединения с последующим процессом внутримолекулярного нуклеофильного присоединения. Во-первых, гидроксид отводит α-водород от концевого углерода с образованием енолята. Затем нуклеофильная атака енолята на другую кетогруппу образует новую углерод-углеродную связь (красная) между атомами углерода 2 и 6. Наконец, обычно в условиях нагревания, удаление молекулы воды дает циклизованный α, β-ненасыщенный кетон.
 Рис. 2: Внутримолекулярная альдольная реакция в полном синтезе (+) - Вортманнина.
Рис. 2: Внутримолекулярная альдольная реакция в полном синтезе (+) - Вортманнина. Внутримолекулярные альдольные реакции широко используются в полном синтезе различных природных продуктов, особенно алкалоидов и стероидов. Примером является применение внутримолекулярной альдольной реакции на стадии замыкания кольца для полного синтеза (+) - вортманнина Shigehisa, et al. (Фигура 2).
Недавняя методология теперь позволяет проводить гораздо более широкий спектр альдольных реакций, часто с каталитическим количеством хирального лиганда. Когда в реакциях используются небольшие количества энантиомерно чистых лигандов для индукции образования энантиомерно чистых продуктов, реакции обычно называют «каталитическими, асимметричными»; например, сейчас доступно множество различных каталитических асимметричных альдольных реакций.
Ключевым ограничением хирального вспомогательного подхода, описанного ранее, является неспособность N-ацетил имидов реагировать избирательно. Ранний подход заключался в использовании временной тиоэфирной группы:
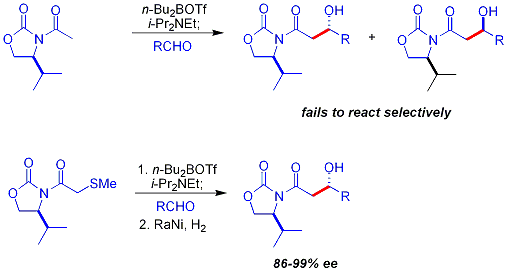
альдольная реакция Мукаямы представляет собой нуклеофильное присоединение простые эфиры силиленола в альдегиды, катализируемые кислотой Льюиса, такие как трифторид бора (как эфират трифторида бора ) или тетрахлорид титана. Альдольная реакция Мукаямы не соответствует модели Циммермана-Тракслера. Каррейра описал особенно полезную асимметричную методологию с силилкетенацеталями, примечательную высоким уровнем энантиоселективности и широким спектром субстратов.
Метод работает с неразветвленными алифатическими альдегидами, которые часто бедны электрофилы для каталитических асимметричных процессов. Это может быть связано с плохой электронной и стерической дифференциацией между их энантиофасами.

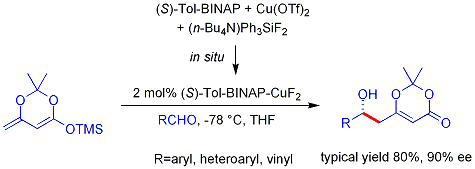
. Аналогичный винилогичный альдольный процесс Мукаяма также может быть сделан каталитическим и асимметричным. Пример, показанный ниже, работает эффективно для ароматических (но не алифатических) альдегидов, и считается, что механизм включает хиральный, связанный с металлом диенолат.
Более поздняя версия вспомогательного вещества Эванса это тиазолидинтион Crimmins . выходы, диастереоселективности и энантиоселективности реакции, как правило, высокие, хотя и не такие высокие, как в сопоставимых случаях Эванса. Однако, в отличие от вспомогательного вещества Эванса, тиазолдинтион может выполнять реакции ацетат-альдола (см. Crimmins, Org. Lett. 2007, 9 (1), 149–152) и может производить «синус Эванса» или «синус не Эванса». аддуктов, просто варьируя количество (-) - спартеина. Считается, что реакция протекает через шестичленные переходные состояния , связанные с титаном,, аналогичные предлагаемым переходным состояниям для вспомогательного вещества Эванса. ПРИМЕЧАНИЕ: в структуре спартеина, показанной ниже, отсутствует атом N.

Более поздней разработкой является использование хиральных вторичных аминных катализаторов. Эти вторичные амины образуют временные енамины при воздействии кетонов, которые могут энантиоселективно реагировать с подходящими альдегидными электрофилами. Амин вступает в реакцию с карбонилом с образованием енамина, енамин действует как енолоподобный нуклеофил, а затем амин выделяется из всего продукта - сам амин является катализатором. Этот метод енаминового катализа является разновидностью органокатализа, поскольку катализатор полностью основан на небольшой органической молекуле. В оригинальном примере пролин эффективно катализирует циклизацию трикетона:
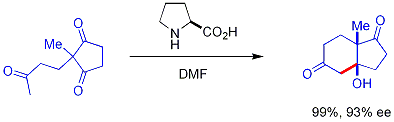
Эта реакция известна как реакция Хаджоса-Пэрриша (также известная как реакция Хаджоса-Пэрриша-Эдера- Реакция Зауэра-Вихерта, ссылаясь на одновременный отчет Шеринга о реакции в более жестких условиях). В условиях Хаджоса-Пэрриша необходимо только каталитическое количество пролина (3 мол.%). Опасности возникновения ахиральной фоновой реакции нет, поскольку промежуточные енаминовые промежуточные соединения гораздо более нуклеофильны, чем их исходные кетонные енолы. Эта стратегия предлагает простой способ создания энантиоселективности в реакциях без использования переходных металлов, которые могут иметь недостатки, связанные с токсичностью или дороговизной.
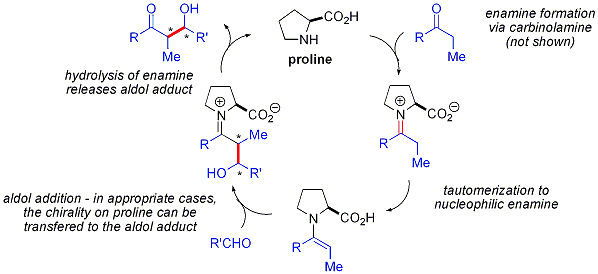
Катализируемые пролином альдольные реакции не проявляют каких-либо нелинейных эффектов (энантиоселективность продуктов прямо пропорциональна энантиочистоте катализатора). В сочетании с изотопной маркировкой доказательствами и вычислительными исследованиями предлагаемый механизм реакции для альдольных реакций, катализируемых пролином, выглядит следующим образом:
Эта стратегия позволяет решать другие сложные задачи. кросс-альдольная реакция между двумя альдегидами. В общем, кросс-альдольные реакции между альдегидами обычно являются сложными, поскольку они могут легко полимеризоваться или реагировать неселективно, давая статистическую смесь продуктов. Первый пример показан ниже:
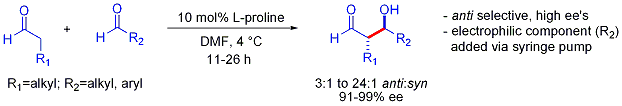
В отличие от предпочтения син-аддуктов, обычно наблюдаемого при добавках альдолов на основе енолятов, эти органокатализируемые добавки альдолов являются антиселективными. Во многих случаях органокаталитические условия достаточно мягкие, чтобы избежать полимеризации. Однако селективность требует медленного добавления желаемого электрофильного партнера, контролируемого шприцевым насосом, поскольку оба реагирующих партнера обычно имеют енолизуемые протоны. Если один альдегид не имеет енолизируемых протонов или альфа- или бета-разветвлений, может быть достигнут дополнительный контроль.
Элегантная демонстрация силы асимметричных органокаталитических альдольных реакций была раскрыта MacMillan и соавторами в 2004 году в их синтезе дифференциально защищенных углеводов. В то время как традиционные методы синтеза обеспечивают синтез гексоз с использованием вариантов итерационных стратегий снятия защиты и снятия защиты, требующих 8–14 шагов, органокатализ может получить доступ ко многим из тех же субстратов, используя эффективный двухэтапный метод. протокол, включающий катализируемую пролином димеризацию альфа-оксиальдегидов с последующей тандемной альдольной циклизацией Мукаямы.
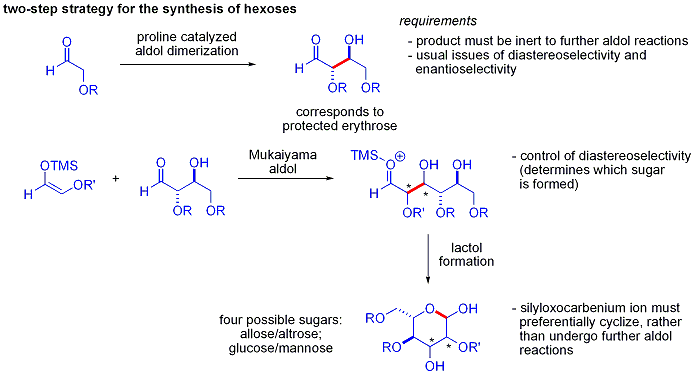
Альдольная димеризация альфа-оксиальдегидов требует, чтобы альдольный аддукт, сам по себе альдегид, был инертен по отношению к дальнейшим альдольным реакциям. Более ранние исследования показали, что для этой реакции подходят альдегиды, содержащие альфа-алкилокси или альфа- силилокси заместители, тогда как альдегиды, содержащие электроноакцепторные группы, такие как acetoxy не реагировали. Защищенный продукт эритрозы затем может быть преобразован в четыре возможных сахара посредством добавления альдола Мукаямы с последующим образованием лактола. Это требует соответствующего диастереоконтроля при добавлении альдола Мукаяма и продукта силилоксикарбениевого иона для предпочтительной циклизации, а не для дальнейшей альдольной реакции. В итоге были синтезированы глюкоза, манноза и аллоза :
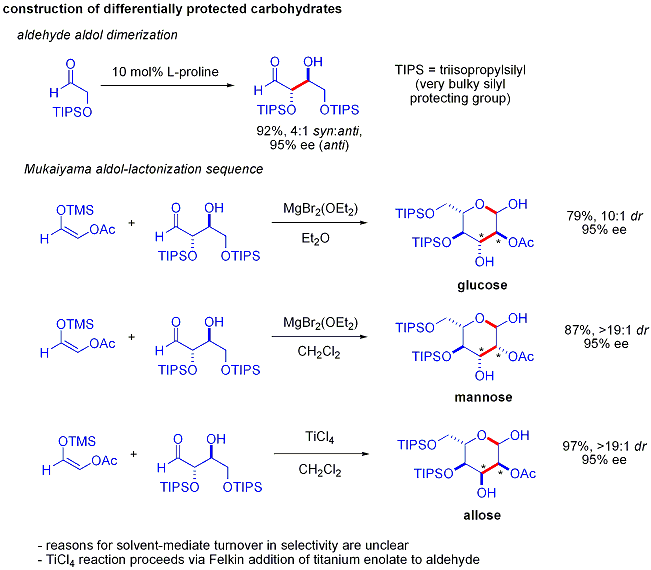
При обычном альдольном добавлении карбонильное соединение депротонируется с образованием енолята. Енолят добавляют к альдегиду или кетону, который образует алкоксид, который затем протонируется при переработке. В принципе, более совершенный метод позволил бы избежать необходимости в многоступенчатой последовательности в пользу «прямой» реакции, которая может быть проведена за одну стадию процесса. Одна из идей состоит в том, чтобы получить енолят с использованием металлического катализатора, который высвобождается после механизма добавления альдола. Общая проблема заключается в том, что при добавлении образуется алкоксид, который является гораздо более основным, чем исходные материалы. Этот продукт прочно связывается с металлом, предотвращая его реакцию с дополнительными карбонильными реагентами.
Один из подходов, продемонстрированный Эвансом, заключается в силилировании альдольного аддукта. В реакцию добавляется кремниевый реагент, такой как TMSCl, который заменяет металл на алкоксиде, обеспечивая оборот металлического катализатора. Сведение к минимуму количества стадий реакции и количества используемых реакционноспособных химикатов приводит к экономичной и промышленно полезной реакции.
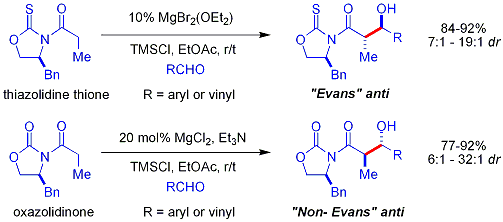
В более позднем биомиметическом подходе Shair в качестве нуклеофила используются бета-тио кетокислоты. Фрагмент кетокислоты является декарбоксилированным in situ. Процесс аналогичен тому, как малонил-КоА используется поликетидсинтазами. хиральный лиганд представляет собой бисоксазолин. Ароматические и разветвленные алифатические альдегиды обычно являются плохими субстратами.
Примеры альдольных реакций в биохимии включают расщепление фруктозо-1,6-бисфосфата на дигидроксиацетон и глицеральдегид-3 -фосфат на четвертой стадии гликолиза, который является примером обратной («ретро») альдольной реакции, катализируемой ферментом альдолазой A (также известной как фруктоза- 1,6-бисфосфатальдолаза).
В глиоксилатном цикле растений и некоторых прокариот изоцитратлиаза производит глиоксилат и сукцинат из изоцитрат. После депротонирования группы ОН изоцитратлиаза расщепляет изоцитрат на четырехуглеродный сукцинат и двухуглеродный глиоксилат в результате реакции альдольного расщепления. Механически это расщепление очень похоже на реакцию гликолиза альдолазой А.